The steroid hormone receptors (SHRs) belong to the nuclear receptor (NR) gene superfamily, which represents a class of ligand-dependent intracellular transcription factors that modulate gene expression in response to lipophilic ligands [1,2]. These receptors play diverse roles in cell differentiation/development, proliferation, and metabolism and are associated with numerous pathologies such as cancer, cardiovascular disease, inflammation, and reproductive abnormalities. Thus, SHRs are broadly implicated in normal physiological development and metabolism and represent therapeutic targets for a wide range of human diseases [3-5]. All the SHRs have a central DNA binding domain (DBD) and a carboxy-terminal ligand-binding domain (LBD) with conserved tertiary structures (Figure 1A). The SHRs also contain an amino-terminal domain (NTD), which is divergent among NR family members. Within the NTD of SHRs lies a powerful activation function, AF1 region, which can activate transcription in a ligand independent fashion. The LBD contains a second activation function, AF2 that maps to a surface-exposed hydrophobic pocket, providing a docking site for coregulatory proteins [6]. Ligand binding to its receptor results in transactivation of specific target genes in a tissue/cell- and promoter- specific manner.
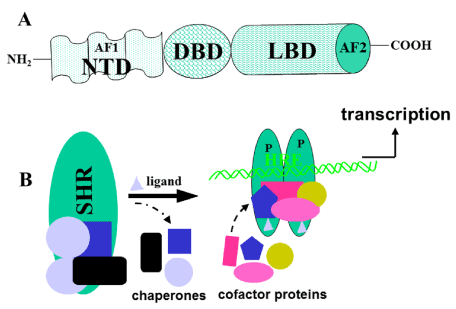
Figure 1. A) A general topology of the SHRs, showing three principal functional domains. The DBD is the most conserved domain and consists of two zinc fingers. It is responsible for site-specific DNA binding at its response element. The second most conserved domain, LBD, is responsible for ligand binding, interaction with heat shock proteins to stabilize receptor, homo and/or hetero-dimerization, and contains a ligand-dependent transcriptional activation region (AF2). Among the members of superfamily, the N-terminal domain (NTD) is the least conserved, both in size and primary sequence, and AF1, lies within this domain. This region is rich in acidic amino acids, and appears to be mostly unstructured. B) Model for the general mechanism of action of SHRs. Without ligand, the receptor (SHR) is present in an inactive complex associated with heat shock and other chaperone proteins. Ligand (gray triangle) binding releases these proteins, hyper-phosphorylates (P) and receptor adopts a conformation that binds with high affinity to its specific DNA response element (HRE) and various cofactor proteins, to regulate transcription. The protein binding partners are shown by different colors and shapes.
Ligands allosterically control the interactions of SHRs with coactivators and corepressors by influencing the conformation of a short helix, referred to as Helix 12 or AF2 (activation function 2), towards the carboxy-terminal end of the LBD. Coactivators bind to the AF2 surface via the amino acid motif LxxLL, in which the Leucine residues dock into the hydrophobic cleft [7,8]. Binding specificity is added by oppositely charged amino acids at either end of the NR hydrophobic cleft that form a charge clamp with the LxxLL peptide backbone. The mechanism of transcriptional activation of SHRs by AF2 is via recruitment of these coactivators, which mediate chromatin remodeling and recruit the basal transcription apparatus. On the other hand, binding of an antagonist to the ligand binding pocket prevents coactivator recruitment to the AF2 via the LxxLL motif. Thus, AF2 provides allosteric control of transcription mediated by its dynamic localization. AF2 is the structural link between a ligand-bound SHR and coactivator, but the details of this communication and its differential regulation by receptor have remained elusive. Several recent structural and functional studies are beginning to shed light on the details of these signaling pathways, facilitating an improved understanding of how small molecule steroid receptor modulators (SRMs) control cofactor recruitment [9-11].
According to the classic mechanism of steroid action a ligand-bound SHR enters the nucleus and binds to its response element DNA site (Figure 1B). This binding allows formation of a transcriptional initiation complex, directly by interactions between AF1 and AF2 and the complex, or indirectly through co-activators. Our understanding of how AF2 functions has been greatly enhanced by the delineation of the basic LBD structure [12,13]. Clinically this phenomenon has extensively been exploited for the development of small molecule SHR modulators (SRMs). However, most of these SRMs exhibit partial agonist or mixed agonist activities that may not be desirable. This undesired effects of SRMs have often been attributed to their current inability to restrict SHR actions to specific organ/gene targets. Though, it has been suggested that the tissue-specific residual activity of SHRs in the presence of SRMs may be mediated via AF1, yet therapeutic targeting of the AF1 has been limited [12]. A major unresolved issue for SHR signaling and transcriptional activities is the nature of cross talk between AF1 and AF2, especially as modulated by SRMs, DNA and other interacting protein partners. This is despite the fact that full receptor activity requires a synergistic effects of both AF1 and AF2. Though it is still not well established whether this association of the AF1 and AF2 is always direct, indirect, or both (Figure 2).
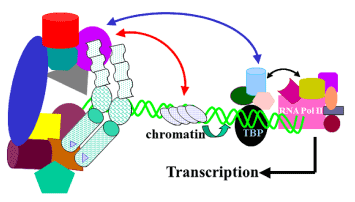
Figure 2. A model for the regulation of transcription by NHR cofactor assemblies. Both AF1 and AF2 regions recruit certain specific cofactors. A bridge is formed between AF1 and AF2 through these and/or other cofactor(s). AF1 and AF2 can also interact directly. Bridging the cofactor(s) are largely regulated by cell-type and ligand. The complex alters local chromatin structure (red arrow), catalyzes histone acetylation or deacetylation, and affects the stabilization of the transcription pre-initiation complex. The receptor complex, bound to DNA enhancer sites, thus recruits and regulates Pol II via direct associations with specific subunits of the mediator complex (blue arrow), which makes a bridge between the receptor and Pol II. The activity of kinases and phosphatases regulating signaling pathways also contribute to this process by altering the state of phosphorylation of both receptor and cofactors (not shown). The receptor:cofactor assembly may also interact directly with the basal transcription machinery at TBP/TATA box to regulate transcription.
Because SHRs regulate many genes in many tissues, synthetic SRMs usually show beneficial therapeutic effects and unwanted side effects that limit their clinical uses. Major goals in the therapeutic targeting of SHRs therefore include attaining a better understanding of the mechanisms underlying their actions in specific cell types and ways in which to selectively modulate their activities [14]. Therefore, importance of SHRs in the regulation of function of all major organ systems demands a full understanding of their mechanism of action. Better understanding SHRs’ inter-domain connectivity should provide the basis for answering several outstanding issues such as the nature of the structural shifts that occur in the SHR upon binding ligand and/or the correct DNA sites in the rapidly reversible interactions between receptor and other proteins, chromatin and DNA sites, and how certain ligands act as agonists in some tissues and antagonists or very weak partial agonists in others. It is anticipated that SHR structure information will ultimately help explain how diverse SRMs and other ligand independent signaling mechanisms are able to elicit selective biological effects, via interactions and/or communication between the AF1, AF2 in diverse tissues. Though the 3-D structures of the independently expressed recombinant DBD and LBD of many SHRs are known, as yet the full structure of none of the member proteins is known. As for the NTD structure is concerned, we are only beginning to understand and there is no 3-D structure of NTD available.
Recent studies have shown that the SHRs’ NTD/AF1 exist as an ensemble of conformers also known as “intrinsically disordered (ID)”, which collectively appear to be unstructured [15-17]. This dynamic unstructured nature of SHR NTDs has been problematic in preventing crystallization and high-resolution atomic structures. Importantly, this problem is not unique to SHRs, and many activation domains of transcription factors contain ID regions that have been refractory to crystallization [18,19]. In recent years, it has become quite evident that these regions of disorder in signaling proteins seem to promote molecular recognition through their ability to form surfaces capable of binding specific binding partners [20]. These dynamic ensembles of interconverting conformers of SHRs’ ID AF1/NTD are capable of undergoing a disorder-to-order transition upon interaction with macromolecules including other proteins or DNA [18,19]. This structural flexibility and process of “coupled folding and binding” appears to have certain advantages for intra- and intermolecular interactions as compared with ordered structural motifs. It has been suggested that the tissue-specific residual activity of SRM-bound SHRs may mainly be mediated via AF1 and that the relative functional importance of AF1 may be decided by specific SRM-induced conformational changes in either LBD or transmitted allosterically to the NTD [12,14]. It therefore makes sense to investigate the possibility of identifying receptor modulators that act to regulate AF1 activity, which could complement or replace existing SRMs. However, the therapeutic targeting of ID NTD/AF1 using small-molecule inhibitors of receptor function has been limited despite the critical role that its structural flexibility can play in allosteric modulation of synergy between AF1 and AF2.
Targeting ID proteins by small molecules to block protein-protein interactions is a rapidly evolving field, and therefore identifying compounds that bind to NTD/AF1 could be promising small molecules for SHR-based therapeutics. A major challenge, though, with developing compounds that target ID NTD of SHRs is the limited knowledge of the physiologically relevant NTD/AF1-interacting coregulatory protein(s) capable of inducing functionally active folded state of NTD/AF1. Meaningful screens for drugs that block interactions of proteins with NTD/AF1s will therefore require identification of the most functionally important coregulatory proteins or protein complexes that interact with the NTD/AF1. Nonetheless, a multifactorial approach of SRMs, coregulators, and assorted small-molecule inhibitors in an organ-dependent context may provide the additional selectivity needed to target selected genes and thereby reduce the number of undesirable side effects in current endocrine-related therapies.
- Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940-954. [Crossref]
- Mangelsdorf DJ, Evans RM (1995) The RXR heterodimers and orphan receptors. Cell 83: 841-850. [Crossref]
- McKenna NJ, O'Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108: 465-474. [Crossref]
- Kumar R, Thompson EB (2012) Folding of the glucocorticoid receptor N-terminal transactivation function: dynamics and regulation. Mol Cell Endocrinol 348: 450-456. [Crossref]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ (2001) Nuclear receptors and lipid physiology: opening the X-files. Science 294: 1866-1870. [Crossref]
- Huang HJ, Norris JD, McDonnell DP (2002) Identification of a negative regulatory surface within estrogen receptor alpha provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol 16: 1778-1792. [Crossref]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95: 927-937.
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, et al. (1998) Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev 12: 3343-3356. [Crossref]
- Johnson AB, O’Malley BW (2012) Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol 348: 430-439. [Crossref]
- Nilsson S, Koehler KF, Gustafsson JÅ (2011) Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov 10: 778-792. [Crossref]
- McDonnell DP, Wardell SE (2010) The molecular mechanisms underlying the pharmacological actions of ER modulators: implications fornew drug discovery in breast cancer. Curr Opin Pharmacol 10: 620–628. [Crossref]
- Kumar R, McEwan IJ (2012) Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation. Endocr Rev 33: 271-299. [Crossref]
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, et al. (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389: 753-758. [Crossref]
- Simons SS Jr, Edwards DP, Kumar R (2014) Minireview: dynamic structures of nuclear hormone receptors: new promises and challenges. Mol Endocrinol 28: 173-182. [Crossref]
- Goswami D, Pascal B, Kumar R, Edwards DP, Griffin PR (2014) Structural dynamics and inter domain crosstalk of PR-TBP interaction probed by hydrogen/deuterium exchange Mass Spectrometry. Structure 22: 961-973. [Crossref]
- Kumar R, Moure CM, Khan SH, Callaway C, Grimm S, et al. (2013) Regulation of the structurally dynamic disordered amino-terminal domain of progesterone receptor by protein induced folding. J Biol Chem 288: 30285-30299. [Crossref]
- Khan SH, Awasthi S, Guo C, Goswami D, Ling J, et al. (2012) Binding of the amino terminal region of coactivator TIF2 to the intrinsically disordered AF1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J Biol Chem 287: 44546-44560. [Crossref]
- Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, et al. (2006) Intrinsic disorder in transcription factors. Biochemistry 45: 6873-6888. [Crossref]
- Dunker AK, Uversky VN (2010) Drugs for ’protein clouds’: targeting intrinsically disordered transcription factors. Curr Opin Pharmacol 10: 782-788. [Crossref]
- Ferreon AC, Ferreon JC, Wright PE, Deniz AA (2013) Modulation of allostery by protein intrinsic disorder. Nature 498: 390-394. [Crossref]
2021 Copyright OAT. All rights reserv