Abstract
This article aims to provide a broad coverage of over 300 studies on T helper 17 (Th17) cells published mainly between 2011 and 2016, with a focus on factors negatively regulating Th17 cell differentiation and functions. During the last decade, processes underlying Th17 cell differentiation and activation, as well as Th17-specific cytokines, chemokines, and transcription factors, have been characterized. Diverse modalities controlling Th17 cells range from factors modulating the state of regulatory T (Treg) cells or dendritic cells and indirectly regulating Th17 cells to cell-intrinsic factors, such as those that repress genes encoding Th17 signature cytokines, including artificial products. Since IL-17 is a major player in tissue-specific immune pathology, Th17 cells, a major source of the cytokine, have been a subject of intensive research and have been at the forefront of clinical studies. New approaches, including conditional knockout mice as well as transcriptome profiling, have revealed closely related developmental states in Th17 cells, reflecting their plasticity. For example, given that Th17 cells share a differentiation pathway with Treg cells that, in turn, control Th17 cells, the Treg/Th17 axis is important for fine-tuning the intensity of inflammatory responses. An emerging picture shows that a combination of many factors involving IL-23, IL-2, CCR6, the mammalian target of rapamycin (mTOR)-hypoxia-inducible factor (HIF) axis, metabolism (glycolysis and lipid synthesis), retinoic acid, glucocorticoids, melatonin, Wnt pathways, and salt act in synergy to regulate the Th17/Treg balance and inter-Th17 subset balance. Therapeutic interventions that can tune such balances would be efficacious when accompanied by our attentiveness to the spatial and temporal dynamics of Th17 cells. A comprehensive understanding of biochemical and cellular factors underlining these subtle regulations would give us a more integrated view that would hopefully help increase therapeutic options for many cases of autoimmune and inflammatory diseases and predisposed individuals.
Introduction - an overview of immunotherapy, IL-17, and Th17 biology
An understanding of the cytokines responsible for autoimmune diseases has changed the concept of their treatment. Prior to the identification of IL-17, several cytokines crucial for autoimmune diseases had been identified. The treatment of autoimmune diseases, including rheumatic arthritis (RA), has been revolutionized by the advent of targeted biological agents as well as improved use of conventional drugs. Inhibitors of TNF-α have shown benefits in many patients with RA and psoriasis [1-3]. This success was followed by the development of drugs targeting IL-6 and IL-1 [2,4,5]. After the discovery and characterization of IL-17A demonstrating that it induces IL-6 secretion from synoviocytes in patients with RA [6], blockade of IL-17A has been assessed in RA, psoriasis, and other related diseases and has shown some successful results [7]. (Henceforth “IL-17” indicates IL-17A, the founding member of the IL-17 family, unless otherwise noted.) Pathological roles of IL-17 in autoimmune diseases, as well as clinical trials targeting IL-17 or IL-23, have been reviewed by Beringer et al. [7], Waisman et al. [8], and Kim et al. [3]. Roles for IL-17 in central nervous system (CNS) diseases, including multiple sclerosis (MS) and infarction [8], and in cardiovascular diseases [9] have also been discussed. To avoid redundancy, we focus on T helper 17 (Th17) biology and mechanisms that inhibit Th17 cell development and activity, with emphasis on their therapeutic relevance. Notably, Th17 is not the only cell capable of producing IL-17; other types of cells, such as γδ-T cells and innate lymphoid cells (ILCs), are likely to be the main source of IL-17 in some cases [10,11].
Upon activation by an antigen, naive CD4+ T cells proliferate and differentiate into various subsets of T helper (Th) cells, including Th1, Th2, and Th17 cells. Th17 cells develop mostly from naive T cells, produce proinflammatory cytokines IL-17A, IL-17F, and IL-22, and coordinate inflammatory responses for host defense [8,12]. Th17 cells have been shown to be important for mucosal host defense against microbial and fungal pathogens [13], but, on the other hand, are present at tissue inflammation sites and contribute to the pathogenesis of human autoimmune and chronic inflammatory disorders [12,14-17]. In mice, Th17 cells express the transcription factor retinoic acid-related orphan receptor γt (RORγt, corresponding to human RORc) as a master transcriptional regulator [18] along with the chemokine receptors CCR6 [19] and CCR2 [20]. Both RORγt and RORα, a closely related family member, are necessary for full Th17 cell development [18]. Multiple cytokines, including TGF-β, IL-6, IL-1β and IL-21 are known to induce differentiation of naive T cells to Th17 cells [12]. In particular, this differentiation can be initiated by a combination of TGF-β and IL-6 in mice [12], and is maintained by IL-23 [21,22]. It is now known that Th17 cells consist of subsets with differential inflammatory potential, ranging from a subset that is induced by TGF-β and IL-6, produces IL-10, and is a weak inducer of inflammation, to a highly inflammatory subset that produces GM-CSF/IFN-γ and is induced by IL-23 [23]. IL-23R is required for effector Th17 cell responses in vivo [24], and IL-23 appears to be a promising therapeutic target [25]. The importance of pleiotropic cytokine TGF-β for Th17 (as well as inducible regulatory T [Treg] cell) development was established by early findings [26]. As the differentiation state of dendritic cells (DCs) has profound effects on Th17 differentiation, the extrinsic effect of TGF-β mediated by TGF-β-signaling in DCs is also important [27].
We do not discuss IL-22, a Th17 signature cytokine, in detail, but instead suggest recent articles [28,29]. Both IL-17 and IL-22 play a central role in the pathogenesis of MS [30,31] and RA [32]. A recently proposed subset, Th22, shows similarity with Th17 [33], yet, one feature of Th22 cells is their dependency on aryl hydrocarbon receptors (AHR), rather than RORγt. Th22 cells play a pathological role in psoriasis, but their role in RA is less clear [34].
Cytokines and other factors present during T cell priming events can direct differentiation by inducing lineage-specifying transcription factors that act as master regulators. T-bet, signal transducer and activator of transcription (STAT)1, and STAT4 are the master regulators for Th1 cells; GATA3 and STAT6 direct the Th2 lineages, and STAT3 and RORγt direct differentiation of Th17 cells [18,35]. The induction of RORγt is dependent on STAT3, which is mainly activated by IL-6. PI3K/AKT signaling acts upstream to positively regulate the activation of protein kinase mammalian target of rapamycin (mTOR)C1, and this axis is a positive regulator of Th17 development [36,37]. mTORC1 positively modulates IL-17 expression through several pathways involving STAT3 and hypoxia-inducible factor 1α (HIF1α) [36,38].
This article focuses on recent reports of negative regulators that inhibit Th17 cell development or suppress their functions, with a limited coverage of the basics revealed in earlier studies. Nonetheless, given the therapeutic relevance of the overall tone of inflammation, and the indirect effect of non-T cells such as DCs, the macroscopic mechanisms governing the balance of various subsets of T cells are also important. Therefore, we begin with Th17/Treg balance, to which we aim to endow an introductory purpose. After the factors modulating Th17 development in an extrinsic manner are discussed, the regulatory factors intrinsic to T cells (i.e., without the aid of other cell types) are considered.
Cytokines, hormones, and vitamins that negatively regulate Th17 cells include retinoic acid [39,40], IFN- β, IL-10 [41,42], IL-27, Th1 and Th2 cytokines, IFN-γ and IL-4 [43,44]. Many of these factors act, at least in part, in an extrinsic manner, i.e., mediated by DCs and other cells. On the other hand, Th17 cell-intrinsic negative regulators include Foxp3 [45], interferon regulatory factor 4 (IRF4)-binding protein (also known as Def6 or SLAT) [46], peroxisome proliferator-activated receptor γ PPAR-γ [47], liver X receptors (LXRs) [48], and STAT5 [49], which we discuss in some depth. Other negative factors include NR2F6 (Ear-2) [50], growth factor independence 1 (Gfi-1) [51], suppressor of cytokine signaling 3 (SOCS3) [52], TNF receptor–associated factor 6 (TRAF6) [53], protein kinase B (PKB)/Akt signals [54], and E26 transformation-specific sequence 1 (Ets-1) [55,56], but we only briefly mention them in related sections, as these may be integrated into some axes. For example, Ets-1 is involved in the Ets-1-IL-2 axis [56].
IL-23 signaling as a target
IL-23 consists of the p40 subunit of IL-12 and an unrelated p19 peptide. There is a consensus that, in the presence of TGF-β IL-6 triggers differentiation of Th17 cells in mice. IL-23 is essential to establish and stabilize the differentiated states of Th17 cells. In support of this view, IL-23 is important in vaccination models [57]. In Khader et al.’s study, despite no involvement of IL-23 in primary resistance to Mycobacterium tuberculosis, vaccination with a defined peptide from M. tuberculosis established persistent IL-17-producing T cells in a manner dependent on IL-23 [57]. Thus-established IL-17-producing T cells, which appeared to accumulate in the lung, allowed accelerated recall response and protection against infection [58].
Setting aside evidence for IL-23 involvement, several studies have elucidated roles of memory Th17 cells in protection from several microbes. Wüthrich et al. showed the importance of Th17 cells in recall responses against several fungi, specifically, Coccidioides posadasii, Histoplasma capsulatam, and Blastomyces dermatitidis [59]. Chen et al. [60] showed the importance of memory Th17 cells established by vaccination for Klebsiella pneumoniae. In that study, Th17, but not IFN-γ was required for broader (i.e., serotype-independent) protection against K. pneumoniae. Using a baboon model of Bordetella pertussis infection, Warfel and Merkel showed the presence of IL-17-producing memory T cells and IFN-γ-producing memory T cells >2 years after infection [61].
Using an experimental autoimmune encephalomyelitis (EAE) model, Haines et al. [25] showed that memory cells were generated from IL-17+RORγt+ precursors, not noncommitted precursors. Compared to the cells on day 8 (after primary immunization), the cells on day 18 showed better Th17 phenotype stability in an IL-23-dependent manner, implying that the time length of primary immunization is critical for stability of the differentiation state of Th17 cells. Short immunization times allowed differentiation into IFN-γ-producing cells in their setting [25]. IL-23 promoted proliferation of memory Th17 cells and upregulated genes required for cell-cycle progression in Th17 cells. IL-23 also induced T-bet and IFN-γ in Th17 cells.
These findings implicated the therapeutic potential of blocking IL-23. Notably, tildrakizumab (MK-3222), a humanized anti-IL23p19 mAb improved psoriasis in a phase IIb randomized, placebo-controlled trial, although adverse effects, including bacterial arthritis, were reported [62].
Tregs, non-pathogenic Th17, and pathogenic Th17
After naive T cells were shown to differentiate into Th17 cells, it was noted that Treg and Th17 cells emerge from an overlapping developmental program (Figure 1) [63]. This close relationship between Treg and Th17 cells, along with the developmental plasticity of Th17 cells, led to recognition that Th17 cells play not only proinflammatory and defensive roles but also have regulatory roles. Treg cells are broadly classified into two groups: nTregs that develop in the thymus and iTregs that are induced in peripheral organs by TGF-β. Thus, both iTreg and Th17 cells can be induced from naive CD4+ T cells in the periphery upon antigen stimulation and exposure to TGF-β. The notion that Treg and Th17 have a reciprocal (mutually exclusive) relationship [39] leads us to surmise that subtle modulations of Th17-iTreg cell balance can exert immense effects on the outcome of therapeutic interventions. Physiologically, switching differentiation between Treg and Th17 cells is mainly regulated by IL-6 [53]. TGF-β signaling alone leads to Foxp3 expression and induction of Treg, but costimulation with IL-6 can suppress Foxp3 and, therefore, release RORγt from inhibition by Foxp3, promoting Th17 development.
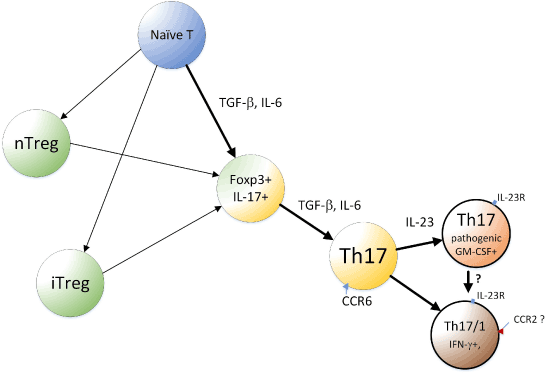
Figure 1. A simplified model of developmental pathway of Th17 and related cells.
Th17 cells can be derived from Treg cells if appropriate conditions are provided. In Veldhoen et al.’s study, in the presence of DC and ligands for TLR3, 4, or 9, coculture of naive CD4+ T cells with Tregs resulted in the development of Th17 cells [64]. In another study, TGF-β likely produced by Tregs and DCs, was a key cytokine that promoted Th17 differentiation from naive CD4 T cells in the presence of dectin-1 agonists [65]. Later, Xu et al. used Foxp3-IRES-GFP knock-in mice, which ensured that only CD4+CD25+Foxp3+ cells were used, and showed that these cells undergo self-induced Th17 differentiation [66]. Treg cells not only expressed TGF-β but also induced DCs to produce increased amounts of TGF-β [66]. A differentiation pathway from the Foxp3+ passing through the Foxp3+/IL-17+ double positive stage and then to the IL-17 single positive stage was also observed [66]. Thus, Treg cells can differentiate into Th17 cells in a manner dependent on TGF-β.
Several studies have focused on the relevance of IL-2 in Th17-iTreg balance. IL-2 is a cytokine that normally suppresses Th17 cell differentiation and function. Later, rather than TGF-β production by Treg cells, IL-2 depletion by Treg cells was proposed to be the key factor promoting early stages in Th17 development. Using an in vivo Candida albicans infection model, Pandiyan et al. found that the effect of Treg cells on the induction of IL-17 production from responding CD4+ T cells is dependent on consumption of IL-2 by Treg cells at early time points [67]. Further, using a system in which diphtheria toxin can kill Foxp3+ Tregs at a specific stage, Chen et al. showed that Treg cells promote Th17 cell development in vivo and this is mediated by consumption of IL-2. Strikingly, their analysis with TGFb knockout mice conditional to Foxp3 expression showed that Treg cell production of TGF-β was not required for Th17 induction in vivo [68]. Cejas et al. showed that TRAF6-deficient mice exhibited enhanced Th17 cell differentiation, and this was at least partly explained by the finding that TRAF6-deficient CD4+ T cells showed lower expression levels of IL-2 compared to those of wild-type CD4+ T cells [53]. Thus, it is possible that negative regulation by IL-2 is playing a central role in suppressing Th17 cell differentiation in many unknown cases. In any case, Pandiyan et al. [67] and Chen et al. [68] showed that Treg cells are likely to promote priming of Th17 in vivo, although further evaluation appears to be necessary to establish the significance of Tregs as a source of TGF-β in vivo. The role of TGF-β in Th17 development is still controversial for human T cells [69]. Effects of IL-2 are also discussed in the following section.
Thus, several findings indicated or suggested derivation of Th17 cells or Foxp3+IL-17+ “double positive” cells from Foxp3+ Treg cells. Of clinical importance, fate-mapping analysis by Komatsu et al. showed that Th17 cells arise from Foxp3+ Treg cells by the loss of Foxp3 expression in the presence of synovial fibroblast-derived IL-6 in collagen-induced arthritis (CIA) model mice, suggesting that T cell plasticity, combined with the inflammatory rheumatic environment, facilitates Th17 polarization, altering the balanced Treg/Th17 ratio [70]. In another study using peripheral blood from patients with RA, Th17 cells were enriched with Helios-producing Foxp3- IL-2RA- cells, suggestive of nTreg cells that had presumably lost suppressive capability [71]. Notably, Helios expression is indicative of the recent thymic origin of the cells, and IL-2RA is abundantly expressed in Treg cells. Thus, in patients with RA, nTreg cells appear to have anomalously high chances of transdifferentiating into IL-17-producing cells.
Ueno et al. observed that the prevalence of circulating double positive (IL-17+ Foxp3+) CD4+ T cells is increased in patients with inflammatory bowel diseases (IBDs) [72]. Basu et al. further showed that IL-1 signaling represses SOCS3, a molecule that normally inhibits STAT3. This was suggested to be the molecular basis for IL-1β-dependent increases in phosphorylated STAT3 and alterations of the STAT3/STAT5 balance resulting in Th17 generation, even in retinoic acid-mediated iTreg induction that is predominant in the normal intestine [63].
It is recognized that Th17 cells have functional plasticity, but can Th17 cells transdifferentiate into Tregs? By using a triple reporter mouse model that reports expression of IL-17A, IL-10, and Foxp3 genes, Gagliani et al. [73] showed that Th17 cells generated during Staphylococcus aureus infection can be converted into IL-10high Foxp3lo Tr-1-like cells. Besides being positive for lymphocyte-activation gene 3 (LAG-3) and negative for CCR6, the latter cells (referred to as Tr-1exTh17 cells) showed features of Tr-1 in transcriptome analyses. TGF-β promoted Th17 to Tr-1 conversion [73]. AHR ligand 6-formylindolo[3,2-b]carbazole (FICZ) also promoted Th17 to Tr-1 cell conversion [73]. Thus, Th17 cells can transdifferentiate into regulatory cells.
Recent notable reports include those of Gaublomme et al. [74] and Wang et al. [75]. Reflecting the functional diversity of Th17, in vitro polarized Th17 cells can either cause severe autoimmune responses upon adaptive transfer (“pathogenic,” polarized with IL-1β + IL-6 + IL-23) or have little or no effect in inducing autoimmune responses (“non-pathogenic,” polarized with TGF-β + IL-6) [76,77]. RNA-seq was performed for single CD4+IL-17+cells isolated in vivo from EAE model mice. Significant cellular variation was observed, and in vivo Th17 showed cell states were progressively changed from the lymph nodes (LNs) to the central nervous system (CNS) [74]. The in vitro Th17 cells were also analyzed after activation under non-pathogenic (TGF-β + IL-6) or pathogenic (IL-1β+ IL-6 + IL-23) conditions. The profiles of these sets of cells formed a spectrum with distinctions and similarities when compared with the profile of the in vivo Th17 [74]. Interestingly, pathogenic Th17 cells expressed T-bet, GM-CSF, and IL-23R, for example, while non-pathogenic Th17 cells expressed IL-10. Exposure of non-pathogenic Th17 cells to IL-23 converted them to a pathogenic phenotype (Figure 1). In humans, Th17 cells that coproduce IL-17 and IFN-γ are generated upon infection with C. albicans, and this state of Th17 appears to be similar to that of pathogenic Th17 cells [75]. Further, in humans, Th17 cells that coproduce IL-17 with IL-10 are induced upon S. aureus infection [78], and this state is more similar to the non-pathogenic Th17 cells [75]. Wang et al. further reported CD5L/AIM expression in non-pathogenic, but not in pathogenic, Th17 cells. CD5L behaved as a functional switch; its loss converted non-pathogenic Th17 cells into pathogenic Th17 cells. CD5L inhibits this conversion in a manner mediated by modulation of the intracellular lipidome, such as maintaining a high ratio of poly-unsaturated fatty acids (PUFA)/saturated fatty acids (SFA) and restriction of cholesterol synthesis, and, thereby, ligand availability of RORγt. Notably, cholesterol synthesis is considered to be linked to the production of RORγt ligands, including oxysterol [79]. Thus, it is reasonable to consider that lipid metabolism plays important roles in T cell-mediated immunity, helping Th17 cells adapt to protective, as well as inflammatory, immune responses.
IL-2
Treg cells are highly dependent on IL-2 for survival, and the number of Treg cells can be dramatically reduced by neutralization of IL-2 [80]. In contrast, Th17 cell responses have been shown to be inhibited by IL-2. As considered above, Chen et al. showed that IL-2 depletion by Tregs acts as a positive regulator in the priming of Th17 cells [68]. In an attempt to utilize the immunosuppressive effect of IL-2, several authors administered low-dose IL-2 treatments in animal models and patients with type I diabetes [81]. To cite a few reports, in an application for type I diabetes therapy/prevention, IL-2 induced a dose-dependent increase in the proportion of Treg cells, without inducing deleterious changes in glucose-metabolism variables [82]. Webster et al. found that the in vivo activity of IL-2 can be enhanced by coinjection of anti-IL-2 mAbs and, intriguingly, one particular IL-2 mAb, when injected into mice as IL-2/anti-IL-2-mAb complexes, selectively expanded Treg cells in many organs [83]. Thus-expanded Treg cells showed excellent suppressive functions, inducing resistance to EAE induction and conferrence of tolerance to islet allografts. Further studies using IL-2/anti-IL-2-mAb complexes include that by Wang et al. that showed that this complex attenuates lung inflammation and heart failure progression in a congestive heart failure mouse model [84].
Ets-1 is a transcription factor belonging to the Ets family and is important in hematopoietic cell development [85]. Moisan et al. showed that Ets-1 is a negative regulator of Th17 development [55]. Ets-1-deficient cells produced less IL-2 than wild-type cells, and Ets-1-deficient mice expressed abnormally high levels of IL-17 in the lung [55]. Tsao et al. showed that Ets-1 promotes IL-2 expression, synergizing with nuclear factor of activated T-cells (NFAT) in the transcription of IL-2 [86].
Zelante et al. focused on a role of IL-2, especially from DCs, to adjust Th17 activity [87]. Using a mouse model of invasive pulmonary aspergillosis, the authors showed that lung CD103+ DCs produce IL-2, leading to an optimally protective Th17 response. Mice conditionally lacking IL-2 in CD11c+ DC cells exhibited unrestrained production of IL-23 and fatal hyperinflammation, which was characterized by the emergence of a Th17 stem-cell-like population [87].
IL-4
Early studies have shown that systemic IL-4 immunotherapy improves Th1/Th17- or Th17-mediated diseases, such as EAE [88], experimental colitis [89], nonobese diabetes [90], and psoriasis in humans [91]. Using several DC populations, Guenova et al. found that IL-4 abolished the capacity of DCs to produce IL-23 while promoting IL-12p70. Further, an IL-4 therapy attenuated Th17-related diseases through STAT6- and activating transcription factor 3 (ATF3)-dependent suppression of the IL-23/Th17 responses, despite simultaneous enhancement of IL-12/Th1 responses [92]. One merit of such cytokines for clinical use would be their long history of research, which could help prevent adverse effects.
STAT3 and a subset of Foxp3+ Tregs regulating Th17 cells
STAT3 is a molecule responsible for programming Th17 effector cells; activation of STAT3 serves as the primary input to the genetic network that governs Th17 differentiation. In an influential study on a subset of Treg cells that regulate Th17, Chaudhry et al. [93] showed that STAT3 expression is important for a subset of Tregs that specifically regulate Th17 cells. Unlike Foxp3 knockout mice that show generalized lymphadenopathy, STAT3-deficient mice conditional to Foxp3 promoter activation showed only splenomegaly and enlargement of the mesenteric lymph nodes, suggesting specific deregulation of Th17, as Th17 cells are mainly located in the intestine. The conditional knockout did not affect the number of Treg cells, but led to a selective increase in Th17 responses. Th1 and Th2 were kept in check by the conditionally STAT3-deficient Treg cells. Intriguingly, their gene expression analysis showed that 20% of Foxp3-dependent genes are also dependent on STAT3 expression in Treg cells.
Koch et al. showed that Th1 immunity is under the control of Th1-specialized Tregs, namely, Treg1 cells, proposing the concept of lineage-specific Tregs [94]. Thus, Th1 and Treg1 share T-bet, and Th17 and Treg17 share STAT3. Key mediators of differentiation signals specific to a helper T cell subgroup are also utilized in a corresponding subgroup of Foxp3+ Tregs, likely endowing unique homeostatic and migratory properties optimized for suppression of the corresponding Th cells. Such examples include CXCR3 in Th1 and Treg1, IRF4 for Th2 and Treg2, and CCR6 in Th17 and Treg17 [94-97]. Thus, a developmentally related Treg subgroup may have been evolutionarily integrated into the homeostasis of each Th subgroup. This could provide benefits to immune system homeostasis as inappropriate distribution of Treg cells in vivo leads to tissue-specific inflammatory disease [98].
Using STAT3-knockout mice conditional to Foxp3 expression, Kluger et al. recently showed that these mice showed increased peritoneal Th17 responses, compared to wild-type controls, after i.p. pristane injection that is known to induce SLE in mice [99]. They reported that the lack of Treg17 cells also caused severe pulmonary vasculitis, as well as, at 4 and 9 months after the injection, aggravation of lupus nephritis, accompanied by enhanced Th17 responses [99]. They also found a reduced level of CCR6 in the Tregs from the conditional KO mice, supporting the CCR6-dependent anti-inflammatory effect of Treg17 cells.
Antigen stimulation and anergy induction
A major part of this article discusses cytokines, reflecting our understanding that pathophysiological Th17 differentiation is profoundly regulated by cytokines. Yet, TCR signaling triggered by self-antigen as causative for autoimmune diseases has long been considered of pathological importance, although it is not easy to identify specific causative antigens in most settings. Using cell transfer analysis between SKG mice that develop autoimmune arthritis (due to the ZAP-70 mutation) and Rag2-knockout mice, Ito et al. presented a method to isolate arthritogenic TCR and revealed that the self-antigen was a ribosomal protein, RPL23A. The presence of anti-RPL23A antibody was confirmed in the serum of 16.8% of patients with RA, signifying the importance of this self-antigen [100]. Yet, although this work seems outstanding, it is generally difficult to determine such antigens due to ethical and technical problems. Even in the case of SKG mice, TCRs on arthritogenic CD4+ T cells were found to be highly polyclonal and varied among individual mice. Is it possible to identify and use peptides to induce Th17 cell tolerance in a TCR-specific manner?
In general, successful vaccination requires targeting antigens to DCs as an appropriate method to stimulate immune responses [101]. As DCs express various receptors on their surface, including TLRs, mannose receptors, and DC-SIGN receptors, targeting such receptors for efficient delivery of antigens has been utilized for efficient anticancer immunotherapy [101]. DCs can take up mannosylated proteins, and present peptide antigen thereof, with a very high efficiency. Different stimulation methods of DCs, and their combinations (in receptor type and strength, for example), can lead to different immune responses. Hawiger et al. [102] showed that in vivo targeting of antigens selectively to steady-state (immature) DCs, by fusing them to an antibody against the DEC-205 endocytosis receptor, induced peripheral T cell tolerance in mice [102]. In Tseveleki et al.’s study, DCs loaded with myelin peptide conjugated to oxidized mannan induced anergy in antigen-specific Th1 and Th17 cells and tolerance in EAE in a transfer analysis [103]. Although further studies are needed to characterize the mechanism for the anergy induction, these findings suggest the potential usefulness of tolerance induction for therapeutic intervention in MS.
B7/CD28 costimulation - unexpected suppression
CD28 was the first costimulatory receptor identified on T cells, and signaling through this receptor usually initiates potent T cell activation [104]. CTLA4 expressed on Treg cells is considered to compete with CD28 on effector T cells for costimulatory ligands and, in the hope of utilizing this effect, treatment with CTLA-4–Ig (abatacept) has been attempted in both RA and psoriatic arthritis [e.g., 105]. Paradoxically, treatment of EAE with Abs against B7 family members (i.e., anti-CD80 or anti-CD86 Abs) or injections of CTLA4-Ig actually exacerbated disease [106]. Moreover, the development of ulcerative colitis during CTLA4-Ig (abatacept) therapy in a patient with RA was reported [107]. These findings led to Bouguermouh et al.’s study showing that CD28 stimulation has a suppressive effect on Th17 cells. In a system with conventional plate-bound anti-CD3 stimulation of mouse peripheral CD4+ T cells under Th17-polarizing conditions, CD28 costimulation decreased the proportion of IL-17-producing cells [108]. CD28 costimulation did not inhibit fully differentiated Th17 cells, but inhibited the polarization of naive CD4+ T cells into Th17 [108]. The inhibitory effect of CD28 stimulation was dependent on IL-2 and IFN-γ, likely secreted by the T cells used (CD4+ T cells) [104]. In support of the inhibition by CD28, coculture with bone marrow-derived DC (BMDC) and CTLA4-Ig showed that interrupting the B7 costimulatory pathway favored Th17 differentiation [108]. This CTLA4-Ig effect can, at least in part, explain the early observation that Tregs facilitate the differentiation of Th17 cells in a proinflammatory cytokine milieu [64,65]. Note, however, that a conflicting result has been reported; Ying et al. argued that addition of human (h)CTLA-Ig, that would mainly block CD28-CD80 interaction, suppressed the production of IL-17, as well as IL-4 and IFN-γ by anti-CD3-stimulated WT CD4+ T cells. When it was applied to CD28-/- CD4 T cells, it enhanced IL-17 production, presumably by blocking CTLA4-CD80 interaction [109]. The cause for this discrepancy between the two studies [108,109] is not clear, but it may be due to differences in population or differentiation stage of T cells used.
Another case in which Tregs can enhance Th17-dependent inflammation has recently been reported by Watanabe et al. [110]. In their system, ovalbumin (OVA) epitope-specific iTregs, Th1, Th2, and Th17 cells were independently prepared in vitro and intravenously transferred to wild-type mice. Various combinations of cotransfer showed that the cotransferred iTregs suppressed Th1- and Th2-mediated colon thickening, but stimulated Th17-mediated colon thickening. Prior oral administration of OVA led to immunosuppression of Th2- and Th1-mediated colon thickening, but instead accelerated Th17-mediated colon thickening. The augmentation by iTregs of Th17-mediated intestinal inflammation depended on CTLA4 [110]. This corroborates Bouguermouh et al.’s results [108] showing an adverse effect of CD28 signaling on Th17 differentiation.
Dendritic cells (DCs) and extrinsic effects of TGF-β
As we have seen above, TGF-β promotes Th17 cell differentiation Veldhoen et al. showed that mice expressing dominant-negative TGF-β receptor (TGF-βR)II showed resistance to EAE through a reduction in Th17 cells [26,64]. The inhibition of the protease thrombospondin-1 (TSP-1), that normally activates TGF-β delayed the onset of EAE [111], in accordance with the role of TGF-β-induced Th17 in EAE. However, TGF-β has long been considered to be an anti-inflammatory factor. In fact, early studies had shown that TGF administration reduced incidence and severity of EAE [112,113] and that injections of anti-TGFβ1 antibody worsened EAE both in incidence and severity [113]. How can we explain such disparity?
DCs can provide a microenvironment suitable for Th17 differentiation. DC development induced by retinoic acid has an impact on Treg and Th17 development, as discussed in the next section. Here, we focus on Speck et al.’s study that used mice with DC-specific knockout of TGF-βR to investigate the role of TGF-β in their EAE model [27]. DCs lacking TGF-β signaling showed a highly mature DC profile and caused severe inflammation and Th17 response in CNS [27]. Using in vitro experiments with bone-marrow precursors, the authors also showed that TGF-β controls (limits) DC numbers at a precursor level, but not at the mature stage. This study is important because, while the promotive effect of TGF-β on Th17 cannot be doubted, extrinsic effects of TGF-β on Th17 are complex, and this careful experimental setting revealed a rather suppressive extrinsic effect of TGF-β via DCs. For efficacious therapeutic intervention in the future, analyses addressing differences due to tissue/compartment, cell type, and timing should be helpful. Complexity of TGF-β effects is likely to manifest as sensitivity to details of intervention protocol such as timing, dose, and methods of delivery chosen for treatment.
Retinoic acid in intestinal mucosal immunity
Retinoic acid, a vitamin A metabolite, plays important roles in embryonic and adult tissue development including immune cells [114]. Retinoic acid promotes the differentiation of iTreg cells, and, in mucosal immunity, fine-tunes the Treg-Th17 balance [115,116]. Retinoic acid generally can: 1) suppress IL-12-mediated Th1 differentiation, 2) enhance IL-4-mediated Th2 response, and 3) enhance TGF-β-induced Treg differentiation by upregulating Foxp3 [114,116].
Retinoic acid potentiates induction of gut-homing Foxp3+ Treg cells, reciprocally inhibiting Th17 cells in vitro. Mucida et al. observed that exogenous retinoic acid inhibited TGF-β- and IL-6-dependent Th17 induction in vivo in an infection model, allowing Treg differentiation, but injection of retinoic acid receptor (RAR) antagonists caused a decrease in Foxp3+Tregs in the lamina propria [39]. Thus, in conjunction with TGF-β, retinoic acid enhances the expression of Foxp3. Analyses of lamina propria by Denning et al. showed that there is normally a high production of cytokine TGF-β and IL-10 [117].
However, retinoic acid is not always immunosuppressive, its effect varying depending on the balance of various cytokines. Physiological concentrations of retinoic acid promote Th17 differentiation in vitro, whereas higher concentrations of retinoic acid inhibit Th17-cell responses in vitro and in vivo [118]. It is also unlikely that the findings in gut analyses can be extrapolated to non-gut phenomena; Pino-Lagos et al. showed that retinoic acid signaling in CD4+ T cells is necessary for T cell tissue accumulation and skin-graft rejection, which represents a role of retinoic acid opposite to tolerance induction [119].
DCs and their expression of retinaldehyde dehydrogenase (RALDH) activity are considered important for the immunological environment of the intestine. Broadly, the majority of retinoic acid functions in immunity are considered to be mediated by canonical RAR/retinoid X receptor (RXR) heterodimers, and by all-trans retinoic acid (ATRA) produced by retinaldehyde dehydrogenase 2 (RALDH2) and acting through RARα [116]. It is noted that most molecular studies highlighted immunosuppressive roles of retinoic acid. Xu et al. showed that retinoic acid stimulation leads to binding of RAR/RXR to the conserved enhancer region (enhancer I), causing increased histone acetylation in the region of the Smad3 binding site and subsequently increased binding of phosphorylated Smad3, thereby leading to Foxp3 expression [120].
In the intestine, DCs, stromal cells, epithelial cells, and macrophages are sources of retinoic acid as shown by high RALDH activity [121,122]. CD103+ DCs in small intestine lamina propria and mesenteric LNs have higher Raldh2 expression relative to DCs in other tissues [123]. Retinoic acid levels correlate with the ability of the intestinal DCs to induce gut-homing potential in T cells [121,124].
Retinoic acid induces T cell homing to mesenteric LNs and gut via the enhanced expression of the gut-homing receptors α4β7 integrin and CCR9 [114,121]. The intestine, lamina propria, and mesenteric LNs have CD103+ DCs that produce TGF-β and retinoic acid, aiding development of Foxp3+ Treg cells. In particular, CD103+ CD11b+ DCs are the most numerous DCs in the small intestinal lamina propria, and are major constituents of the tolerogenic CD103+ DC population [125]. Although the impact of this subset (CD103+CD11b+) on Treg cells is difficult to isolate because of functional redundancy with CD103+CD11b- DCs, a decrease in intestinal Treg cells was observed in animals lacking all CD103+ intestinal DCs [126]. Many recent studies have focused on the effect of retinoic acid on differentiation and modulation of DCs. As an example, Klebanoff et al. showed that mice deprived of retinoic acid signaling show selective loss of splenic endothelial cell-specific adhesion molecule (Esam)high CD11b+ cells that are developmentally related to the small intestine lamina propria CD103+CD11b+ DC subset [127]. Transferred pre-DCs differentiated into the CD11b+ CD8a- subset, but transfer into vitamin A-deficient hosts caused differentiation to the CD11b+CD8a+ lineage [127].
Retinoic acid production and signaling in DCs can be enhanced by many factors, including retinoic acid itself [123], TLR signaling [128], GM-CSF [129], and IL-4 [129,130]. TLR2 signaling in DCs, in particular, appears to be important for maintaining host-microbiota mutualism [128]. Wang et al. showed that signaling of TLR1/2 induces retinoic acid-producing activity in splenic DCs, conferring the ability to imprint T cells for gut-homing [131].
Direct effects of retinoic acid on T cells have also been studied. Lu et al. showed that all-trans retinoic acid (ATRA) increased histone methylation and acetylation within the region including the promoter of Foxp3a, thereby promoting TGF-β-induced Treg development [132]. Nguyen et al. showed that retinoic acid treatment enhances TLR2-dependent IL-10 production by T cells and this, in turn, potentiates Treg cell generation [133]. Round et al. showed that symbiosis factor (PSA) from Bacteroides fragilis can induce Foxp3+, IL-10-secreting Treg cells via TLR2 expressed on CD4+ T cells, promoting immunological tolerance and colonization of B. fragilis on mucosal surfaces [134]. More cases showing synergy with other cytokines and signals for retinoic acid are likely to emerge in the near future.
Unlike vitamin D, whose in-serum level can be measured as 25-hydroxyvitamin D (25(OH)D), serum retinol level does not reflect the intracellular retinoic acid concentration, making clinical studies difficult. Nonetheless, several studies focused on the clinical relevance of retinoic acid. In a mouse model of allergic airway inflammation, ATRA treatment attenuated airway inflammation and decreased Th2- and Th17-related transcription factors [135]. In vitro analyses also showed that ATRA modified Treg/Th17 balance in favor of Treg cells [135]. Vitamin A level (measured as the retinol binding protein (RBP)/transthyretin (TTR) ratio) was negatively correlated with CD4+ T cell proliferation in patients with MS [136], and vitamin A supplementation in patients with MS upregulated TGF-β and Foxp3 expression in PBMCs [137]. Therapeutic modulation of RALDH activity appears to be a worthwhile approach. Manicassamy et al. [138] showed that Wnt/β-catenin signaling in intestinal DCs is required for Raldh1 and Raldh2 expression, as well as IL-10 and TGF-β and that β-catenin deletion in DCs led to lower Treg and higher Th1/Th17 cell differentiation making the host susceptible to inflammatory bowel diseases (IBD) [138].
Some studies have focused on retinoic acid receptors. Of note, RXRs (RXRα, RXRβ, and RXRγ) can form homo- as well as heterodimers with several nuclear receptors including RARs, vitamin D receptor (VDR), LXR, and PPARγ. CD4+ T cells mainly express RXRα. ATRA binds to RAR and 9-cis–retinoic acid can bind to both RAR and RXR [139]. Recently, Chandraratna et al. showed that RXR activation by IRX4204 promotes iTreg formation, inhibits Th17 development, and can profoundly alleviate disease in EAE mouse models [140]. Brown et al. used mice carrying a dominant negative form of RARα and showed that loss of retinoic acid signaling in fully committed Th1 cells leads to transdifferentiation to cells that have features of Th17 lineage, implying that retinoic acid stabilizes the differentiation state of Th1 and suppresses Th17 pathways [141]. This suppression was necessary to prevent pathogenic Th17-biased responses in Listeria monocytogenes infection and OVA-specific TCR model mice. Retinoic acid-RARα antagonized the activity of transcription factors important for Th17 differentiation (IRF4, basic leucine zipper transcription factor (BATF), STAT3, and RORγt), without any signs of antagonizing Th2-cell-associated genes [141]. Taken together, RALDH, retinoic acid, and its receptors could be a target for therapeutic intervention for the suppression of Th17 cells. It is likely that in the near future further progress will be made in various subareas of vitamin A research.
Vitamin D
Vitamin D, or specifically, its active form 1,25(OH)2-cholecalciferol (1,25-(OH)2D3) is known for its role in maintaining systemic levels of calcium and phosphate. 1,25-(OH)2D3 is derived from 25(OH)D by CYP27B1-mediated reactions and exerts its effects through VDRs. 25(OH)D is commonly used as a marker for vitamin D status. The 1,25-(OH)2D3-VDR-RXR complex recruits coactivator or corepressor complexes, depending on the type of cell, thereby determining the transcriptional response from vitamin D response element (VDRE)-containing genes [142]. Several studies reported a negative correlation between circulating 25(OH)D and MS disease activity [e.g., 143]. In patients with MS with low vitamin D levels, an intervention with supplementary vitamin D significantly reduced disease progression [144]. However, the evidence for vitamin D as a treatment for MS is inconclusive, as all studies were underpowered due to small sample sizes [145]. Notably, Pozuelo-Moyano et al. suggested stratifying by HLA-DR15 (MS risk allele) status in future trials and including patients with progressive forms of MS, considering that the possible protective effect of vitamin D could be masked by subgroups of nonresponders.
Correlations between VDR alleles and MS susceptibility have been reported [146,147], yet no association with VDR alleles was observed in a genome-wide association study despite the identification of 57 regions significantly associated with MS, suggesting weak or no appreciable association of VDR alleles [148]. For several specific alleles of VDR, functional impacts have been shown [142]. Although CYP27B1 mutations are rare and do not contribute a genetic risk in the majority of disease cases, an analysis using an in vitro system allowing differentiation of DCs demonstrated that the risk allele of CYP27B1 is underexpressed in tolerizing DCs [149]. Overall, while genetic evidence for vitamin D involvement in autoimmune pathogenesis is increasing, it seems challenging to pinpoint the causative genes, due to the limited resolution hampering dissection of linkage disequilibrium blocks [149]. There could be multiple causative alleles with weak predisposing effects.
More unequivocal results on the protective effects of vitamin D have been obtained in rodent models. Th1, Th2, and Th17 cells were all shown to express Vdr transcripts and can be modulated by vitamin D [150]. 1,25-(OH)2D3 is likely to have no impact on fully differentiated rodent CD4+Foxp3+ T cells [151], although VDR-dependent downregulation and upregulation of Foxp3 transcription in rodent CD4+Foxp3+ T cells have also been reported [142]. Mixed bone marrow chimera studies showed that 1,25-(OH)2D3 inhibited EAE induction in a manner dependent on Vdr expression in hematopoietic cells [151]. 1,25-(OH)2D3 failed to inhibit EAE induction in mice with knockout of Vdr specific to CD4+ T cells [151]. On the other hand, mice with global knockout of Vdr had Th17 cells that overproduced IL-17 and a reduced number of tolerogenic DC cells [152]. These results support the suppressive role of vitamin D against Th17 functions.
Using cells from patients with hereditary vitamin D–resistant rickets (HDVRR), Tiosano et al. showed that TNF-α and IL-17 concentrations were significantly higher in HVDRR lymphocyte cultures than in controls. 25(OH)D suppressed IL-17 only in control, and not in HVDRR, lymphocytes [153]. However, as the authors discuss, the cohort of 35 patients with HVDRR did not show a higher incidence for infectious or autoimmune diseases, suggesting the presence of compensatory mechanisms that protect patients with HVDRR from such diseases.
Smad3 is a member of the Smad family that transmits TGF-β signaling [154]. Smad3 interacts with the VDR and promotes its function in transcriptional regulation. A recent study by Nanduri et al. showed that 1,25-(OH)2D3-VDR-RXR heterodimer directly binds to the VDRE in the Smad7 promoter and inhibits its expression in Th17 cells [155]. Notably, Smad7 is known as one of the inhibitory Smads that negatively regulate TGF-β signaling in a feedback regulatory mechanism [156]. The VDR axis also activated extracellular signal-regulated kinase (ERK), inhibiting expression of Th17-specific genes.
Both animal and human studies have implicated 1,25-(OH)2D3 as a positive regulator of genes encoding receptor activator of nuclear factor kappa-B ligand (RANKL) and of cutaneous IL-10-producing iTreg cell induction, suggesting the usefulness of phototherapy during periods of light starvation for immune-mediated diseases [142,157]. Overall, however, vitamin D and human Th17 studies have so far provided conflicting data [142]. One relatively unnoticed confounding factor could be melatonin, a hormone that controls circadian rhythms and is immunosuppressive, as we discuss below. Specifically, it could be that the melatonin level is high in winter, assisting the maintenance of an immunotolerant environment, complementing the light starvation in winter that would lower the level of 1,25-(OH)2D3. In general, besides the direct effects on Th17, extrinsic effects mediated by Treg and other immune cells appear important for Th17 regulation by vitamin D.
STAT3 vs STAT1
STAT3 is the primary input to the genetic network that governs Th17 differentiation [158]. The effect of IL-6 on Th17 differentiation is mediated by JAKs that in turn activate STAT members, including STAT3. In contrast, STAT1 reacts negatively to Th17 differentiation. IL-27 is considered to be a potent inhibitor of Th17 differentiation and exerts its inhibitory effect in a STAT-1-dependent manner [159].
However, it was puzzling that IL-10 and IL-27, neither of which supports Th17 differentiation, can activate STAT3. Do other factors enable differential functions of the latter cytokines [158]? Recently, Peters et al. showed that, in STAT1-knockout mice, IL-27 “induced” Th17, suggesting that the ratio of activated STAT3/activated STAT1 is important for the Th17 differentiation program [158].
Hirahara et al. performed genome-wide transcriptome analysis [160]. Intriguingly, there was an extensive overlap of the transcriptomes induced by IL-6 and (Th17-inhibiting) IL-27 with few examples expressed in an opposite manner by the cytokines. They further showed that STAT3 is responsible for the overall transcriptional output for IL-6 and IL-27, whereas STAT1 shapes the specific signature superimposed upon STAT3’s action, driving specificity. Hirahara et al. found that much of STAT1 binding to chromatin is dependent on STAT3, and most likely on heterodimer formation with STAT3.
Targeting RORγt
RORγt, the master transcription factor of Th17 cells, is indispensable for Th17 cell development, but not for other T-helper-cell lineages. RORγt not only promotes the production of IL-17 and IL-22 from Th17 cells, but also promotes these cytokines’ production by lymphoid tissue inducer (LTi) cells and other RORγt+ innate lymphocytes (ILCs), suggesting parallel functionalities of ILCs and Th17 cells during host defense [161]. Notwithstanding the presence of these RORγt+ cells other than Th17, RORγt remains an attractive pharmacologic target for the treatment of Th17 cell-mediated immune disorders. Taking advantage of the ligand-binding pocket of RORγt, small-molecular weight compounds have been screened.
Digoxin has been shown to specifically inhibit the transcriptional activity of RORγt and suppress Th17 differentiation [162]. Digoxin showed no effect on CD4 T cell differentiation toward Th1 and no binding to RORα [162,163]. However, concentrations of 1-2 µM appear to be necessary in vitro, and collagen-induced arthritis (CIA) model mice required 2 mg/kg via thrice weekly injections. This concentration is high, calling for a careful assessment of adverse effects if therapeutic translation is conducted.
Another example is SR1001, a derivative of T0901317 that is an LXR ligand [164]. SR1001 was devoid of all LXR activity yet retained its ability to suppress the activity of RORα and RORγt. Xiao et al. identified three RORγt inhibitors (TMP778, TMP920, and GSK805) that suppress Th17 development and alleviate EAE in model mice [165]. Another molecule that inhibits RORγt is ursolic acid, a small molecule used in herbal medicine [166].
Interestingly, Lin et al. [166] discussed CD4 aptamer-RORγt short hairpin RNA (shRNA) chimeras that enabled CD4-specific shRNA delivery to suppress RORγt expression. To derive the aptamer part of the chimera, systematic in vitro ligand evolution was performed and target-specific aptamers were isolated from a random sequence oligonucleotide library. This technique may enhance the efficacy of in vivo injection of shRNA recombinant plasmid DNA [e.g., 167]
Foxo1
Members of the forkhead box O (Foxo) family of transcription factors regulate many facets of cell physiology, including cell proliferation [168]. Phosphorylation by Akt downregulates their activity. Foxo members are important for specifying T cell differentiation, particularly in the pathway to Tregs [168]. It is still a matter of debate whether Foxo1 directly controls Foxp3 [168], but Foxo1 is likely a T cell-intrinsic regulator of tolerance [169]. T cell-specific deletion of Foxo1 caused spontaneous T cell activation and induction of inflammatory bowel diseases in a transfer model. Foxo1-deficient T cells had low IL-7R expression, compromising IL-7-induced T cell survival and proliferation, and in particular naive T cell homeostasis [169]. Foxo1 acts as a direct repressor of IL-23R expression [170]; the Il23r promoter is transactivated by RORγt in IL-23-restimulated Th17 cells and can be inhibited by Foxo1 [170]. Importantly, using a bone marrow chimera strategy, Laine et al. showed that Foxo1 does not need Tregs to negatively regulate Th17 cell differentiation, and this regulation depends on the direct binding of Foxo1 to RORγt [171]. Given the T cell-intrinsic suppressive activity of Foxo1, it is hoped that in the near future further analyses of this molecule may produce useful findings.
IL-35
IL-35 is an immunoregulatory cytokine that belongs to the IL-12 family and consists of the Epstein-Barr virus-induced gene 3 (EBI3) and the p35 subunit of IL-12 [172]. Notably, IL-27 and IL-35 share the EBI3 subunit. IL-35 is normally produced by CD4+Foxp3+ Tregs and iTr35, a regulatory T-cell population [173]. We only briefly discuss IL-35 here, given the excellent review articles available [e.g., 174,175].
IL-35 has shown its therapeutic effectiveness in several studies [175]. In Niedbala et al.’s study, recombinant IL-35 fusion protein (EBI3-p35-Fc fusion) suppressed CIA, at least in part, by increasing IL-10 in serum and suppressing Th17 cells [176]. In the latter system, IFN-γ was enhanced. Wirtz et al. showed that, compared with IL-27p28-deficient mice, EBI-3-deficient mice showed more severe features of colitis with increased numbers of T cells producing Th1 and Th17 cytokines in mucosa [177]. Recombinant IL-35 suppressed the colitis and reduced levels of systemic markers for Th1 and Th17 cells. Bettini et al. generated NOD transgenic mice in which IL-35 was expressed in pancreatic β-cells. This expression protected the NOD mice from autoimmune diabetes [178]. Kochetkova et al. found that IL-35 suppressed CIA in a mouse model and that CD39+CD4+ regulatory T cell expansion, a new subset of regulatory T cells, plays an important role in this protection [179]. It is possible that IL-35 effects are strongly dependent on IL-10 [179,180].
However, some studies showed proinflammatory functions of IL-35. In Thiolat et al.’s study [180], direct DNA injection followed by in situ electrotransfer into the cells of CIA mice unexpectedly aggravated the CIA and produced increased Th17/Treg ratios. Filková et al. showed an increased level of IL-35 in synovial tissue of patients with RA [181]. In both PBMCs and synovial fibroblasts in RA, in vitro stimulation with TNF-α-induced expression of IL-35. Strikingly, IL-35 treatment increased proinflammatory cytokine production by human PBMCs [181]. Cao et al. showed an enhanced IL-35 level in patients with sepsis as well as in cecal ligation and puncture (CLP)-induced sepsis in mice [182]. In the latter model, a blocking analysis showed that the IL-35 effect is generally suppressive against neutrophil recruitment and proinflammatory cytokine production, but that IL-35 facilitates bacterial dissemination.
IL-35 can activate both STAT1 and STAT4, but this balance is determined by the receptors [174]. STAT1 activation negatively effects Th17 differentiation, but STAT4 is controversial. While the findings of Varikuti et al. suggest that STAT4 is important for Th1 and Th2, but not for Th17 [183], McWilliams’ data suggested that Th17 differentiation is influenced by STAT4 activation [184]. Dulek et al. also showed that STAT4 deficiency strongly affected Th1, but it also affected Th2 or Th17 responses [185].
T cell factor 1 (TCF-1) and Wnt
The Wnt proteins are a large family of palmitoylated secreted glycoproteins that regulate many processes, including cell development. Wnt signaling is important in T cell development as well as in cellular developmental processes and tumorigenesis. Review articles, including that by Ma et al. [186], generally suggest its immunosuppressive role. Among the (at least three) Wnt signaling pathways, a transcriptional coactivator β-catenin serves in the canonical Wnt signaling pathway, by interacting with transcription factors including TCF. Wnt/β-catenin signaling influences T cell polarization in favor of Th2 over Th1 [187] and potentiates the survival of nTregs [188]. Consistent with this, specific deletion of β-catenin in DCs led to low Treg and high Th1/Th17 differentiation [138].
As its name suggests, TCF-1 plays an important role in T cell development, involving transition from the CD4-CD8- double negative to the CD4+CD8+ double positive (DP) state [189]. Ma et al. showed that TCF-1-deficient mice were susceptible to Th17-dependent EAE induction and had a higher proportion of Th17 cells compared to wild-type mice [190]. They observed that TCF-1 regulates Th17 differentiation affecting neither TGF-β-induced Treg differentiation nor expression of Th17 master factors like RORγt, STAT3, RORα, AHR [191], Runx1 [192], Ets-1, IRF4, or BATF. TCF-1 did not appear to affect Th17 differentiation at the mature T cell stage. Rather, analysis of histone acetylation and methylation states suggested that knockout of TCF-1 leads to opening up of the IL-17 locus in the thymus due to chromatin modifications. Thus, TCF-1 is likely to repress IL-17 gene expression via epigenetic modifications during T cell development [190], and is therefore important for the early stage of Th17 development.
Ye et al. compared CD4+ T cell transcriptomes between patients with RA and healthy controls using microarray analysis and pathway analysis. Differentially expressed (DE) genes showed enrichment of immune response, T-cell response, and apoptotic signals [193]. The Wnt signaling pathway showed differential expression; the degree of enrichment of DE genes of STAT3 signaling and that of Wnt signaling were comparable, suggesting the importance of the Wnt signaling pathway in pathological roles of CD4+ T cells in RA development.
Frizzleds are seven-pass transmembrane proteins similar to G-protein-coupled receptors and are the main proteins responsible for binding to Wnt on the plasma membrane. Secreted frizzled-related proteins (sFRPs) make up the largest family of Wnt inhibitors. sFRP1 functionalities involve inhibition of Wnt signaling by hindering Wnt binding to Frizzled and by forming nonfunctional complexes with Frizzled [194]. Lee et al. showed that sFRP1 potentiates IL-17 production from restimulated human memory CD4+ T cells and promotes Th17 differentiation in a manner mediated by inhibition of the Wnt pathway. sFRP1 enhanced TGF-β activity in human T cells, appearing to positively regulate Th17 differentiation [195]. They also showed that sFRP1 and IL-17 levels were high relative to those of patients with osteoarthritis and were positively correlated in the synovial fluid of RA.
TIGIT confers Treg cells suppressive ability specific to Th1 and Th17
T cell Ig and immunoreceptor tyrosine-based inhibition motif (ITIM) domain (TIGIT), also known as VSTM3, is a recently identified coinhibitory receptor that is found on NK cells, memory T cells, follicular Th cells, and on a subset of Tregs [196]. Similar to the well-known competition between CTLA-4 and CD28, TIGIT (on Tregs) and CD226 (on NK, Th1, and CD8+ T cells) share ligands (e.g., CD155 on DCs). TIGIT is considered to have dual (or more) pathways to inhibit T cell responses. Besides the hindering CD226-CD155 interaction, engagement of TIGIT inhibits T cell responses via its cytoplasmic ITIM motifs and recruitment of the phosphatase SHIP-1. Interestingly, TIGIT+ Tregs selectively suppress Th1 and Th17 responses, sparing Th2 responses [197]. Compared to TIGIT-deficient Treg cells, TIGIT+ Treg cells abundantly express CTLA-4, CD25, IL-10, Foxp3, and fibrinogen-like protein 2 (Fgl2), signifying their high suppressive potency. Both soluble TIGIT administration and lentivirus-mediated expression of TIGIT ameliorated CIA in mouse models [198,199]. TIGIT ligation induces secretion of Fgl2, which is necessary for the potent suppression of Th1 and Th17, as well as for prevention of Th2 suppression [197]. Intriguingly, both in vivo and in vitro findings supported a view that IL-17 promoted production of Fgl2 in spleen cells, suggesting the presence of a homeostatic regulation loop [200]. Specific mechanisms for Fgl2 action await further elucidation.
IRF4, BATF and IRF4-binding protein (Def6) and ROCK2
Although we cover only a few studies on transcription factors, a complex picture of the gene network governing Th17 differentiation emerges even when focusing only on proteins associated with IRF4. IRF4, a member of the IRF family of transcription factors, is a key regulator of Th17 development, although its expression is also regulated in other T cells [46]. Irf4-/- mice show a lack of Th17 differentiation and are resistant to autoimmune diseases in models of EAE and colitis. Def6 acts as an inhibitor of IRF4. Def6-deficient mice developed a systemic lupus-like syndrome [201]. In Chen et al.’s study, Def6-deficient mice crossed to DO11.10 mice that carry a transgenic TCR recognizing a specific peptide exhibited spontaneous development of arthritis, large-vessel vasculitis, and enhanced production of IL-17 and IL-21 [202]. They also showed that Def6 sequesters IRF4 and prevents it from targeting the transcriptional regulatory regions of Il-17 and Il-21 [202]. Def6 can directly interact with IRF4 and prevent Rho-associated coiled-coil-containing protein kinase 2 (ROCK2)-mediated IRF4 activation (phosphorylation) necessary for binding to regulatory regions of IL-17 and IL-21 [202,203]. Notably, ROCK2 activation also plays an important role in Th17 differentiation, and Def6 generally acts to suppress ROCK2 activity [203]. Of practical importance, oral administration of the ROCK2 inhibitor KD025 in healthy humans downregulated the ability of PBMCs to secrete IL-21 and IL-17 upon stimulation [204]. KD025 and siRNA-mediated inhibition of ROCK2 negatively regulated STAT3 phosphorylation and reduced the levels of IRF4 and RORγt [204]. Treatment with KD025 successfully ameliorated chronic graft versus host disease (GVHD) in several mouse models [205].
Schraml et al. showed that the activating protein 1 (AP-1) transcription factor, BATF, is required for Th17 development [206]. BATF was found to bind conserved intergenic elements in the Il-17a-Il-17f locus and to the Il-17, Il-21, and Il-22 promoters [206]. Chromatin immunoprecipitation (ChIP) analysis by Glasmacher et al. showed that, in Th17 cells, IRF4 targets sequences enriched for AP-1-IRF composite elements (AICEs) that are cobound by BATF [207]. Thus, both IRF4 and BATF are necessary for Th17 differentiation.
Is there any inhibitory factor against BATF useful for Th17 suppression? Miao et al. [208] showed that early growth response gene (Egr)-2, a zinc-finger transcription factor, interacts with BATF in CD4+ T cells and suppresses its interaction with the Il17 promoter. Conditional Egr-2 knockout did not change the levels of STAT3 activation or RORγt expression. Thus, inhibition of BATF is a unique function of Egr-2, and the BATF pathway appears to be independent of STAT3 and RORγt. In Zhu et al.’s study [209], analysis of mice with Egr-2 knockout conditional to T cells showed hyperresponsiveness in response to TCR stimulation and Th1/Th17 bias resulting in lupus-like syndrome.
Among BATF-associated molecules, IRF8 is notable as a key factor regulating differentiation between Th1 and Th17 pathways. IRF8 can bind to BATF and negatively regulate Th17 differentiation [210]. Retinoic acid upregulates Irf8 in Th1 differentiating cells, thereby suppressing Th17 cell genes [141].
Metabolic pathways and mTOR
Upon activation, T cells dramatically alter their metabolic activity to meet the increased metabolic demands for cell proliferation and effector functions. A number of studies have focused on the system integrating T cell activation and control of their metabolic state [211]. For instance, Chang et al. demonstrated that the effector function of T cells is coupled with the state of glucose metabolism [212]. They first showed that T cells grown with galactose, but not with glucose, shift to a state in which respiration (oxidative phosphorylation) is used, but aerobic glycolysis is not used in the main, for energy acquirement (i.e., generation of ATP). This finding is consistent with earlier reports, e.g., Rossignol [213]. Strikingly, T cells cultured in galactose had a severe defect in IFN-γ and IL-2 production. These findings indicated that the state utilizing aerobic glycolysis is coupled to cytokine production. In the absence of aerobic glycolysis, IFN-γ translation was blocked by enhanced GAPDH binding to IFN-γ mRNA [212]. Thus, engagement/disengagement of aerobic glycolysis allows the post-translational regulation of IFN-γ in T cells. Such coupling may ensure that, when T cells undergo antigen-driven proliferation during the immune response, effector cytokines are produced to meet their requirement; whereas when T cells undergo homeostatic proliferation, and such cytokine production is not necessary, they do not produce cytokines. Increased expression of PD-1 was also shown in the T cells cultured with galactose. Coculture experiments showed that the presence of tumor cells causes nutrient restriction to T cells that dampens cytokine production and, at the same time, increases PD-1 expression [212].
Multiple metabolic programs are controlled by mTOR signaling. After treatment with the mTOR-specific inhibitor rapamycin, effector T cell development was greatly diminished [211]. TCR signaling induces uptake of amino acids including leucine, which is important for activation of mTORC1 and metabolic reprogramming of T cells [214]. Notable studies on metabolism and Th17 development include that of Nakaya et al. showing that stimulation of naive CD4+ T cells via TCRs and CD28 triggers the uptake of glutamine, the most abundant amino acid in plasma [215]. The amino acid transporter ASCT2, that was found to be crucial for glutamine uptake, was required for naive T-cell differentiation to Th1 and Th17 cells, but not to Th2 cells. Amino acids can activate mTORC1 by targeting it to lysosomal membranes for activation [216,217]. ASCT2 was required for TCR and CD28-stimulated activation of mTORC1 pathways, but not for the activation of several other T-cell activation pathways, including the MAP kinase pathways [215].
mTORC1 increases expression of HIF1α [218]; HIF1α in turn, upregulates Glut1 expression increasing glucose uptake [219]. HIF1α was shown to be critical for the development of Th17 in both mice and humans [220,221]. Expression of the enzymes mediating rate-limiting steps in glycolysis are positively regulated by HIF1α [220]. T cells deficient in HIF1α showed impaired ability of Th17 cell differentiation, and this was explained by the decreased glycolysis [220]. Inhibition of glycolysis shifted the Th17/Treg balance in favor of Treg cells. Thus, the tuning of Th17/Treg balance is coupled to a very fundamental biochemical process (glycolysis); this is not specific to Th17, but shared at least by T cells [222]. These findings suggest that TCR- and CD28-stimulation promotes Th17 cells to an enhanced state in which, even in inflamed tissues that are inevitably hypoxic, Th17 cells can utilize glycolysis to generate ATP under the limited oxygen availability, as well as exert effector functions.
Liver kinase B1 (LKB1) is a serine threonine kinase identified as the tumor suppressor responsible for Peutz-Jeghers syndrome [223]. LKB1 is known to activate AMP-activated protein kinase (AMPK), which, in turn, suppresses mTOR activity [224]. As AMPK is considered a conserved guardian of cellular energy [225], there appeared to be an anticipation that LKB1-deficiency and AMPK-deficiency would lead to enhanced activation of Th17 cells. Using LKB1-knockout mice conditional to T cells, MacIver et al. reported that T cells lacking LKB1 show increased rates of glucose uptake and glycolytic activities, as well as enhanced expression of Glut1 [226]. LKB1-deficient T cells showed elevated IFN-γ and IL-17A production, as well as enhanced differentiation toward Th1 and Th17 lineages, relative to control T cells [226]. They further showed that AMPKα1-deficient T cells showed elevated glycolytic activities, but this deficiency did not show an effect on Th1, Th2, and Th17 effector cell differentiation and functions [226]. This unexpected result may represent the common challenge in this type of study: in genetic analyses using knockout and conditional knockout animals, the effect may become masked by compensation [227].
β-oxidation of FA produces copious acetyl-CoA that serves as a fuel for mitochondrial oxidation. Although acetyl-CoA can be derived from pyruvate (the glycolysis product), mitochondrial oxidative metabolism, in theory, can operate if acetyl-CoA is abundant even if pyruvate is not abundant. In general, it has been proposed that the two reciprocal processes (FA synthesis vs FA oxidation) are biased in favor of FA oxidation in iTreg cells, whereas FA synthesis is more important in activated Th17 cells to meet proliferation requirements [228]. This hypothesis postulates that Treg cells rely on mitochondrial FA oxidation to proliferate and this bias is, at least in part, caused by activation of AMPK [228].
Acetyl-CoA carboxylases (ACCs) catalyze the conversion of acetyl-CoA to malonyl-CoA, the key step for regulation of cellular FA metabolism. In both mice and humans ACCs have two isoforms, ACC1 (cytosolic) and ACC2 (associated with the outer membrane of mitochondria) [229]. Using ACC1–knockout mice conditional to T cells and soraphen A, an ACC1 and ACC2 inhibitor, Berod et al. showed that de novo FA synthesis controls the Th17/Treg differentiation pathway. Naive CD4+ T cells cultured under the Th17 polarizing condition in the presence of soraphen A exhibited reduced expression of Il-17f, Stat3, and Hif1a, and enhanced phosphorylation levels of AMPK, all indicative of Treg/Th17 balance modulation in favor of Treg. However, this effect of ACC inhibition is not specific to Th17 lineages; it also suppressed proliferation of CD4+ T cells cultured under Th1- and Th2-polarizing conditions [230].
PPARγ and LXRs - transrepression
Besides its roles in lipid metabolism, PPARγ exerts an anti-inflammatory response in murine and human macrophages [231]. This effect is likely mediated mainly by transrepression; liganded PPARγ inhibits the inflammatory activities of AP-1, STAT-1, NF-κB, and NFAT. This property of PPARγ enables it to promote Th2 cytokine production, decreasing Th1 cytokine production [232].
Using CD4+ T cell-specific PPARγ-knockout mice, Klotz et al. indicated that PPARγ serves as a specific brake of Th17 differentiation. Treatment with the PPARγ agonist pioglitazone (PIO) suppressed Th17 differentiation, but showed no influence on Th1, Th2, or Treg differentiation [47]. Direct interaction of PPARγ with RORγt was not observed. Rather, PPARγ activation caused persistent binding of a corepressor, silencing mediator for retinoid and thyroid hormone receptors (SMRT), to the RORγt promoter. Notably, a ChIP assay showed that clearance of SMRT from the RORγt promoter normally precedes RORγt expression induced by TGF-β/IL-6 stimulation of CD4+ T cells. Thus, anti-inflammatory actions of PPARγ ligands have much to do with stabilization of the repression state of NCoR1/SMRT [233]. Both NCoR1 and SMRT repress proinflammatory genes in macrophages, most of which are normally upregulated by NF-κB. The anti-inflammatory effect of PPARγ ligands involves prevention of NCoR1 dismissal [234]. Anti-inflammatory mechanisms of both PPAR and LXRs involve ligand-dependent self-sumoylation, which in turn inhibits NCoRI ubiquitination or phosphorylation induced by LPS, modifications normally leading to dismissal from promoters in proinflammatory conditions.
LXRs belong to the nuclear receptor superfamily that plays important roles in cholesterol and fatty acid metabolism, positively regulating genes for cholesterol efflux and bile acid synthesis [235]. LXRs have been shown to repress inflammatory gene expression in macrophages [234,236]. Both LXR isoforms (LXR-α and -β) are expressed in CD4+ T cells [237]. Intriguingly, Cui et al. [48] showed that LXRs mediate negative effects on Th17 differentiation. Analysis of mice deficient for both LXR-α and -β showed that LXR protein itself acts to inhibit Th17 differentiation and initiation of EAE. Treatment with LXR agonists (GW3965 and T0901317) decreased expression of RORγt and profoundly inhibited Th17 differentiation. They further identified the E-box element, a putative Srebp-1-binding site, within the IL-17 promoter and found that it is necessary for T0901317-dependent suppression of Th17 differentiation. They also showed that Srebp-1, whose gene is regulated by LXR, suppresses Th17 differentiation by binding the E-box element, thereby binding to and inhibiting the AHR that normally increases Il-17 transcription.
Reflecting their involvement in many cellular processes, LXRs have multifaceted roles in immunity. In murine macrophages, LXR activation antagonizes the NF-κB pathway, thereby inhibiting TLR4-mediated LPS responses [236]. Reciprocal inhibition between TLRs and LXRs has been discussed [238]. However, in human macrophages LXR can potentiate LPS-induced responses [239]. In Korf et al.’s study, treatment with a synthetic LXR agonist showed increased resistance to M. tuberculosis infection resulting from activation of LXR-signaling pathways, that was accompanied by increased Th1/Th17 function in the lungs [240]. This is reminiscent of Joseph et al. [236] that showed the requirement of LXRs for normal immunity to L. monocytogenes. Given such broader implications, assessment of many aspects of metabolism and immunity, as well as detailed evaluations regarding timing and dose, may become important if LXR-based therapeutics are pursued for Th17 suppression. Regarding PPARγ, further regulatory factors may also be elucidated in the near future. For example, T cell expression of epidermal fatty acid-binding protein (E-FABP) promotes Th17 differentiation, while counterregulating development of Foxp3+ Tregs [241]. E-FABPs may act to sequester PPAR ligands in the cytoplasm, thereby inhibiting nuclear entry and PPAR activity [241].
A lipidome-based approach may provide a useful insight into integrate diverse aspects of Th17 development. As discussed above, newly described CD5L behaves as a functional switch; CD5L stabilizes non-pathogenic Th17 cells, inhibiting their change into pathogenic Th17 cells. This inhibition is mediated by intracellular lipidome modulation, such as maintenance of a high PUFA/SFA ratio and restriction of cholesterol synthesis, and, thereby, RORγt ligand availability [75]. From a biological perspective, little is known about the evolutionary advantages of such crosstalk between lipid metabolism and T cell development/functions.
Glucocorticoid and GILZ
Glucocorticoids (GCs) are the most widely used anti-inflammatory and immunomodulatory agents. Most physiological and pharmacological effects of natural and synthetic GCs involve activation of the glucocorticoid receptor (GR). The efforts to separate therapeutic from adverse effects of GCs led to the use of a protein induced by GCs as a drug that may mediate their anti-inflammatory effects [242]. GC-induced leucine zipper (GILZ) is a protein discovered during such efforts and has been suggested to be a key player in the anti-inflammatory activity of GCs [243]. GILZ is widely expressed in various cells, including lymphocytes, DCs, macrophages, and epithelial cells. General mechanisms for GC- and GILZ-mediated transrepression and transactivation have been discussed in Hoppstädter and Kiemer [244]. Using in vitro analyses, GILZ-deficient mice, and skin biopsy samples from patients with psoriasis, Jones et al. showed that GILZ plays suppressive roles against Th17 cells but is downregulated in patients with psoriasis [245].
Mesenchymal stromal (stem) cells (MSCs) deploy various modalities to suppress inflammation [246]. Luz-Crawford et al. showed that MSCs from wild-type, but not from GILZ knockout mice, have immunosuppressive potential when transferred into a CIA murine model. GILZ expression in MSCs was required for the generation of IL-10-producing regulatory Th17 cells [247].
IL-27 and Interferon (IFN)-β
IL-27 was initially described as an initiator of Th1 responses, but was later shown to exhibit ability to antagonize various T cells involving Th17 and to promote Treg cell responses [248]. We discussed IL-27 effects on Th17 cell development in section for STAT3 vs STAT1. Sweeney et al. provided in vivo and in vitro data suggesting that IFN-β exerts its therapeutic effects in patients with MS partly via the induction of IL-27, implicating IL-27 as an alternative therapy for patients with MS that do not respond to IFN-β [249]. In murine models of EAE, both Th1 and Th17 myelin oligodendrocyte glycoprotein-specific T cells were shown to induce EAE with similar severity, but resulted in different anatomical pathologies [250]. In a Th17-biased EAE mouse model, IL-27 suppressed EAE in an IL-10-independent manner [251]. This stands in contrast to the requirement of IL-10 for suppression of Th1-biased EAE by IL-27 [252]. When human naive CD4+ T cells cultured in Th17-polarizing conditions were treated with IL-27 or IFN-β, IL-17 production was reduced to 50% or less relative to the control. Intriguingly, neutralizing anti-IL-10 Ab did not show effects on IL-27 and IFN-β, signifying the non-IL-10-mediated nature of the suppressive effect of IL-27 and IFN-β against Th17 [251].
Conflicting results in serum level in patients with MS have been reported. Tang et al. showed that patients with progressive MS had decreased plasma and mRNA expression levels of IL-27 [253]. On the other hand, in Naderi et al.’s study, plasma levels of IL-27 in patients with MS were increased significantly compared to the control subjects [254].
Melatonin, NFIL3, and ERK1/2
Intriguingly, melatonin, a hormone whose rhythmic production serves as an important day-night and seasonal endocrine signal, is linked to Th17-suppressive transcription factor nuclear factor, interleukin 3 regulated (NFIL3). NFIL3 (also known as E4BP4) is a basic leucine zipper transcription factor that has been shown to have an association with IBDs [255]. Yu et al. [256] showed that Nfil3-/- mice had higher IL-17A+ and RORγt+ Th17 cell frequencies than wild type mice in both small intestine and colon. NFIL3 suppresses Th17 cell development by direct binding and repression of the RORγt promoter [256]. A nuclear receptor, REV-ERBα is involved in the transcriptional network of the circadian clock and directly represses Nfil3 transcription, thereby linking Th17 cell development to the circadian clock network [256,257]. Perturbed light-cycles caused increased Th17 cell frequencies in the intestine and spleen of mice in a manner dependent on the expression of REV-ERBα [256].
A cohort study by Farez et al. [258] demonstrated seasonality of MS relapses, specifically, a 32% reduction in the number of MS relapses occurring during fall and winter, in accordance with Jin et al. [259] and Spelman et al. [260]. Treatment with melatonin ameliorates disease in an EAE mouse model [258]. Under Th17-polarizing conditions, IL-17, but not IFN-γ production from human and murine CD4 cells was suppressed by melatonin. Farez et al. further showed that melatonin induces expression of NFIL3 and activation of ERK1/2, which is known to suppress Th17 [258,261]. Mechanistic details of melatonin signaling involving REV-ERBα have been shown by Farez et al. Unsurprisingly, melatonin effects are not specific to Th17; melatonin treatment induced Tr1 differentiation via ERK1/2 and RORα [258].
Targeting salt effects
Intriguing reports from Kleinewietfeld et al. and Wu et al. showed that mouse naive CD4+ T cells cultured in high-salt (sodium chloride) medium had a higher expression of serum/glucocorticoid-regulated kinase 1 (SGK-1) and produced higher numbers of Th17 cells compared to those cultured in normal conditions [262,263]. This effect was mediated by the p38/MAPK-NFAT5 pathway [262]. Loss of SGK1 abrogated Na(+)-mediated Th17 differentiation in an IL-23-dependent manner [263]. Hernandez et al. observed that in a mouse model of MS, a high-salt diet exacerbated disease progression and impaired Treg function [264]. In an EAE mouse model, Jörg et al. suggested that a direct effect of NaCl on Th17 cells, rather than an effect primarily exerted via DCs, plays the key role [265]. We direct readers to a recent review by [266].
Epidemiological studies were also informative. In a cross-sectional analysis using a detailed questionnaire on 18555 individuals including 392 (self-reported) patients with RA, a logistic regression model showed that the odds for RA increased with daily sodium intake. A logistic multivariate model adjusted for many confounding factors (including age, sex, prevalent cardiovascular diseases, diabetes, and smoking) showed that the fourth quartile had an odds ratio of 1.5 (P < 0.02) [267]. Their case-control study replicated the dose-dependent association. Farez-Fiol et al. recently showed that sodium intake is associated with increased disease activity in MS [268]. Note, however, that McDonald observed no strong association between dietary salt intake and pediatric-onset MS risk [269].
In Monteleone et al.’s study, in vitro analysis using human lamina propria mononuclear cells showed enhanced expression of IL-17, IL-23R, TNF-α and RORγt following NaCl exposure in a p38/MAPK-dependent manner, while expression of IFN-γ was unchanged. In vivo analyses of mice fed a high-salt diet showed a consistent result [270]. Recent intriguing reports include that from Wen et al. Analyses with human subjects, as well as in vitro experiments with Jurkat cells, showed that potassium supplementation has a blocking effect on IL-17A production in T lymphocytes induced by salt loading. This protective effect was found to be mediated by the direct suppression of the p38/MAPK-SGK1 pathway [271].
Dopamine as a target
Several findings have suggested immunological effects of dopamine or its antagonists. This is interesting, because dopamine has long been therapeutically utilized, mainly as a neurotransmitter, although expression in many organs, including the gastrointestinal tract, is recognized [272]. As the comprehensive review by Levite discusses, expression of dopamine receptors (DR) on T cells, with differential balance among DR types depending on T cell subsets and stages, influences Th1/Th2/Th17 differentiation [272]. At least five DRs are known (D1R to D5R), all being G-protein coupled receptors. D1R/D5R couple with stimulatory Gα subunits, while the remaining receptors couple with inhibitory Gαs [273]. In many settings, dopamine suppresses Treg in an autocrine or paracrine manner, thereby enhancing effector T cell activities. Several studies support the view that dopamine also directly stimulates effector T cells. In some cases, anomalous expression of DR, or responses to dopamine, is reported for MS, RA, and SLE. Given the article by Levite [272], we cite only a few recent papers.
Using D5R knockout mice, Prado et al. showed that D5Rs expressed on DCs are able to modulate the development of T cells and, in particular, activate differentiation of Th17 cells. D5R-deficient DCs transferred into wild-type recipients reduced the severity of EAE [273,274]. However, conflicting results exist among analyses of patients. In Ferreira et al.’s study, PBMCs from patients with relapsing-remitting (RR)-MS showed increased proliferation and production of TNF-α, IL-6, and IL-17 upon stimulation with phytohemaglutinin (PHA) + dopamine, relative to cells from healthy controls [275]. Further, IL-17 and IL-6 production by T cells from patients with MS was less sensitive to glucocorticoid inhibition, supporting the idea that dopamine stimulates Th17 proliferation. However, in Melnikov et al.’s study [276], patients in the relapse stage of MS exhibited a high level of IL-17 and, intriguingly, lower level of dopamine in serum relative to healthy controls and patients in the remission stage. In that setting, dopamine treatment mildly reduced IL-17 in PBMC cultures from the relapse patients, although after the dopamine treatment IL-17 levels remained high relative to those of PBMC from patients in remission. It is not clear why dopamine promoted Th17 proliferation in Ferreira et al.’s study [275], but inhibited it in Melnikov et al.’s [276], yet Melnikov et al.’s finding of high IL-17 production after dopamine treatment of PBMCs from patients with RR-MS is consistent with that in Giorelli et al.’s study [277]. However, while these results reinforce the role of Th17 cells in MS activity [278], Melnikov et al.’s results represent a challenge in a dopamine-centered view of MS pathogenesis [276].
Gut microbiota, SCFA, and histone deacetylase (HDAC) inhibitors
Intestinal dysbiosis (microbial imbalance) has been shown to be associated with, or suggested to contribute to, the pathogenesis of various autoimmune diseases, including both IBD and non-IBD [279-284]. The importance of gut commensal bacteria in regulating the Treg/Th17 axis has been widely recognized [285]. Although recent studies showed that gut microbiota remotely regulated systemic disease by driving the induction and egress of T follicular helper (Tfh) cells of gut Peyer’s patch [e.g., 286], here we briefly discuss microbiota relevance to Th17 cells.
Atarashi et al. discovered several strains of Clostridia that promote expansion of Treg cells and, upon oral administration, attenuate colitis and allergic diarrhea in mouse models [287,288]. Although they are minor among gut microbiota, mucosa-associated species such as segmented filamentous bacteria (SFB) can powerfully modulate host immunity [289]. SFB adhesion to small intestine epithelial cells leads to induction of Th17 [290,291]. In the K/BxN TCR transgenic mouse model of inflammatory arthritis used by Wu et al., Th17 cells were essentially absent from the site in germ-free K/BxN mice but monocolonization of SFB was capable of triggering arthritis development in K/BxN mice through promotion of Th17 cell populations in the small intestine lamina propria and spleen [289]. Intriguingly, Kumar et al. recently showed that IL-17 receptor (IL-17R)-deficient mice exhibited an earlier onset and worsened severity in an EAE model. They also used IL-17R knockout mice conditional to enteric epithelium and found dysbiosis of SFB (SFB overgrowth), increased serum GM-CSF concentrations, enhanced predisposition to neuroinflammation, and unconstrained Th17 development [292].
Findings derived from many studies led to the consensus that symbiosis between Tregs and commensal microbes is important. In particular, Kawamoto et al. provided findings arguing that mucosal IgAs, diversified and selected in a manner dependent on Foxp3+ T cells, contribute to the maintenance of diversified and balanced microbiota, which in turn facilitate the expansion of Foxp3+ T cells, induction of germinal centers, and IgA responses in the gut, comprising a symbiotic regulatory loop [293]. Short chain fatty acids (SCFAs) and in particular, butyrate, have been shown to facilitate acetylation of the Foxp3 histone upregulating this gene, thereby promoting iTreg development [294,295]. Mechanistic insights were gained by the discovery of G-protein-coupled receptors (GPCRs) for SCFAs, which mediate their anti-inflammatory action [296].
For gut bacteria to play an integral role in maintaining intestinal immunity against pathogens and tolerance against self-antigens, metabolites from gut bacteria, including SCFA, are considered important. SCFA shows inhibitory activity against HDAC [297,298]. Koenen et al. reported a profound negative effect of the HDAC inhibitor trichostatin A on the emergence of IL-17-producing cells from Tregs, implying that Treg differentiation into Th17 cells depends on histone deacetylase activity [299]. In general, histone acetylation often accompanies gene transcription, required for the appropriate tissue-specific induction of many genes, and is opposed by the activity of HDAC [132]. This is consistent with the view that, in the case of Tregs, histone acetylation-mediated upregulation of Foxp3 expression may be important for stabilizing Treg status. Indeed, histone H3 and H4 acetylation was found to be associated with upregulation of Foxp3, which is important for Treg differentiation [132,300]. HDAC9 knockout mice showed increased numbers of Treg cells with increased suppressive capacity [300]. HDAC inhibitors (HDACi) are classified into two groups; those of Class II enhance the suppressive function of murine Treg cells, while Class I HDACi have shown mixed results [298].
The story of commensal bacteria, SCFA, and HDAC is illuminative, but HDACi treatment may face a challenge for therapeutic translation, as is often the case with interventions in intracellular phenomena that are often ubiquitous. The inhibitory effects of HDACi on effector CD4+ T cells are absent or very weak after activation of these cells [298]. HDACi also enhance the function of cytotoxic CD8+ T cells. Therefore, timing and procedure of treatment may need to be carefully optimized in approaches to modulate ubiquitous events. Yet, HDACi appears to have a good chance of efficacy in many pre-clinical and clinical settings, particularly when a short-term treatment can be critical. For example, Sugimoto et al. showed in an islet transplantation mouse model in which donor-specific blood perfusion (DST), when combined with HDACi treatment, dramatically improved graft survival [301]. This effect was accompanied by a pronounced decrease in IL-17 mRNA levels in the spleen and Treg cell induction in the thymus.
IL-25 (IL-17E)
IL-25 (also known as IL-17E) belongs to the IL-17 family, but, unlike IL-17A, IL-25 promotes Th2-type immune responses, contributing to atopic dermatitis and asthma [302]. IL-25 exhibits anti-inflammatory properties in many settings where IL-17 is involved [303]. IL-25-deficient mice are susceptible to EAE, exhibiting increased IL-23 levels and a subsequent increase in inflammatory cytokines involving IL-17 [304]. Several findings suggest that IL-25 is of particular importance in commensal bacteria-dependent induction of tolerance in the intestinal immune system. Notably, the number of Th17 cells in the large intestine increases (>3 fold) in the absence of commensal bacteria [305]. Zaph et al. also showed commensal-dependent expression of IL-25 by intestinal epithelial cells and that this inhibits macrophage production of IL-23, thereby limiting Th17 proliferation [305]. Intriguingly, IL-25 did not suppress Il12a, Tgfb, Il6, or Il10 gene expression, showing specific negative regulation of IL-23 by IL-25. Later, Su et al. showed that IL-25 is markedly decreased in the sera and mucosa of patients with IBD and that IL-25 normally inhibits CD4 T-cell activation and differentiation into Th1/Th17 cells in an IL-10-dependent manner [306].
IL-17RA is likely to the most important receptor mediating the effects of IL-17A [307]. When IL-17RA is considered as a therapeutic target to reduce IL-17A effects, however, the outcome could be confounded by the IL-25 effect, as the biological activities of IL-25 require both IL-17RB and IL-17RA [303]. Unlike anti-IL-17A inhibitors that showed efficacy in patients with RA [308], trials using anti-IL-17RA (brodalumab) in patients with RA showed no efficacy [7,309,310]. This suggests that IL-17RA targeting abrogates the IL-25 effect. IL-25 (IL-17E) signaling may have to be retained during therapeutic intervention. On the other hand, when it comes to the therapeutic potential of IL-25 itself in patients with IBD, evaluation of adverse respiratory system events may become important [311]. This motivates us to seriously think about locally acting biologics not mediated by systemic circulation.
Aryl hydrocarbon receptor (AHR)
There are many other molecules and mechanisms regulating Th17, although we cannot exhaust them all here. AHR is important in T cell differentiation and function, although it was only briefly discussed above (in sections for "Tregs, non-pathogenic Th17, and pathogenic Th17"and "PPARg and LXRs). AHR-deficient mice have increased levels of Th17 and IL-17/IL-22-producing γδT cells in a skin-inflammation model [312]. However, in vitro studies suggest that AHR paradoxically promotes Th17 differentiation [313]. Various endogenous ligands with apparently differential effects on Treg/Th17 balance are present [314], and expression patterns of AHRs are broad and complex [191]. Broadly, there are two conflicting views. In Nguyen et al.’s study, AHR ligation in DCs is required for full Treg cell differentiation [315]. On the other hand, Stephens et al. showed that inhibition of kynurenin 3-monooxygenase that catabolizes kynurenin, an AHR ligand, in Th17 cells caused increased IL-17 production in vitro [316]. Further analyses of kynurenin, a tryptophan metabolite, and AHR activation in Th17 are warranted. It is generally difficult to predict the outcome of a therapeutic approach targeting AHR [191]. AHR has also been targeted in the context of ILCs. AHR is required for the development of RORγt+ ILCs and the production of IL-22 from these cells [317]. AHR is also required for cryptopatches (CPs) and isolated lymphoid follicles (ILFs) in the intestine, with ILFs being a sort of inducible tertiary lymphoid organ in the intestine that develop from the CPs, (rudimentary lymphoid structures) [161,317].
Other modalities
Adenosine acts as an immunosuppressive molecule [246]. The role of the CD39/adenosine axis may have been underevaluated in Th17-mediated diseases. CD39 is an ectoenzyme that catalyzes the conversion of ATP to 5′-AMP, the substrate for CD73. CD73 catalyzes production of adenosine from 5′-AMP [246]. Importantly, in patients with juvenile autoimmune liver disease, Th17CD39+ cells are markedly diminished and fail to generate AMP/adenosine, compared to healthy subjects, thereby limiting control of both target cell proliferation and IL-17 production [318]. In the near future, involvement of the CD39/adenosine axis in other diseases may also be elucidated.
miRNAs (micro-RNAs) are short fragments of non-coding RNA that bind to the 3ʹ-UTR of complimentary mRNA, thereby repressing/silencing target RNAs [319,320]. miRNAs are important for the regulation of Th17 development and functions, but we do not cover many studies here, as there are excellent review articles [320,321]. The pathological roles and diagnostic potential of miRNAs in RA have been reviewed by Churov et al. [322]. miRNA roles in the pathogenesis of MS and IBD have been discussed by Wu and Chen [323] and Xu et al. [324], respectively. From the perspective of negative regulation of Th17 cells, miR-210 acts as a negative regulator of Th17 differentiation. Deletion of miR-210 promotes Th17 differentiation under hypoxic conditions [325]. Other miRNAs regulating Th17 cells include miR10, miR210, miR301, and miR326 [321]. miRNA’s role in Treg and Th17 regulation is clearly important, but this area is still in its infancy [321]. In fact, besides the miRs discussed by Ueno et al., Naghavian et al. showed that miR141 and miR200a are likely to be key miRNAs in the progression of MS through differentiation of Th17 cells and inhibition of differentiation of Treg cells [326]. In the coming years, more information on miRNAs relevant to Th17 will be elucidated through preclinical and clinical studies.
Perez et al.’s analysis using siRNA showed that lymphocytes (mouse and human) externalize calpains, calcium-activated proteases generally considered to act in cytosol. Extracellular calpains negatively regulated IL-17A production by lymphocytes by cleaving TLR2, and thereby preventing lymphocytes responding to TLR2 ligands. They further showed that low-dose IL-2 increased calpain secretion and decreased lymphocyte expression of TLR2, and that this decrease was abolished by calpastatin. Using peritonitis and autoimmune arthritis models, they further showed in vivo relevance of calpain externalization in regulating (reducing) IL-17A expression [327].
Several authors have suggested or indicated the negative effect of the antioxidant CoQ10 on inflammation and Th17 cells. Tawfik showed that intraperitoneal administration of CoQ10 not only potentiated the antiarthritic effect of methotrexate (MTX) but also alleviated MTX-induced hepatocellular injury [328]. Oral administration of CoQ10 ameliorated zymosan-induced arthritis in mice [329]. Splenocytes from CoQ10 treated mice showed decreases in IL-17, CCL20, and RORγt mRNA levels, and an increase in Foxp3+ Treg cells [329].
Although the mechanistic explanation has yet to be given, Bcl2 family molecules have drawn attention in the pathogenesis of autoimmune disease. The Bcl2 family consists of three classes of proteins that can either promote or inhibit apoptosis [330]. B cell lymphoma 2–interacting (Bcl2-interacting) mediator (Bim) is considered to delete autoreactive lymphocytes through apoptosis. Paradoxically, Bim-deficient mice showed protection against the development of EAE and diabetes [330]. Other paradoxical reports related to Bcl-2 involve that of Iglesias et al. who showed that mice with T cell-specific overexpression of BCL2A1, an antiapoptotic Bcl2 family member, had attenuated development of CIA. Both in vivo and vitro, Th17 differentiation was impaired. In vitro TCR stimulation showed defective activation of p38 MAPK [331]. Characterization of the BCL2A1 interactome may reveal a novel pathway regulating the p38MAPK pathway [331].
Although we do not cover the subject broadly, several cases have been reported in which MSCs inhibit Th17 cell activity. Luz-Crawford et al. showed that MSCs inhibit Th1 activity in a manner dependent on soluble factors, but MSC-suppression of Th17 differentiation was mediated by direct contact [332]. Using antibodies to PD-L1 and PD-1, they showed that PD-L signaling was the key for this suppression [332]. Other studies that discussed PD-1-mediated suppression include that by Yang et al. who showed that PD-1-deficient mice develop severe CIA. When T cells from CIA mice were analyzed ex vivo, mice lacking PD-1 exhibited aberrant antigen-specific Th17 responses. Deregulated activation of PKC-θ and Akt was suggested as the cause for these aberrant responses [333].
Perspective
As we have seen above, some cytokines, including IL-2 and IL-23, showed robust negative regulation in many settings. However, for several factors, including TGF-β, AHR signaling, vitamin D, IL-35, IL-27, and dopamine, the effects depend on the cell type and complexity arising from interactions between non-T cells and Th17 cells, and their spatiotemporal dynamics appear to cause significant dependency of effects of these factors on timing, dose, location, and context.
Fine-tuning the balance between regulatory cells and Th17 cells is important. The same is true for pathogenic and non-pathogenic subsets of Th17 cells. Local imbalance, likely in the intestine, between such populations causing over-proliferation of Th17 cells results in exacerbation of autoimmunity in remote organs [99]. It is reasonable to interpret that necessity of the tight regulation of the balance favored the pairwise evolution of Th17 and Th17-targeted Treg cells sharing a developmental pathway and forming clusters in tissues. It is likely that, in the early phase of development, Th17 cells located in the vicinity of Tregs receive promotive stimuli from Tregs (TGF-β, IL-2 depletion, and CTLA-4 mediated suppression of CD28 signaling). Such time- and stage-sensitive roles of Tregs on Th17 cells might have evolved in such a way that Tregs enable a near-optimum time-course of Th17 functionality in protection against microbes, and at the same time, avoidance of autoimmune diseases. Such pair-wise evolution, which would enable evolution of well-coordinated dynamics, is also seen in cytokines. IL-17 families are basically proinflammatory, but IL-25 (IL-17E) exerts suppressive effects on Th17 in the IBD model, despite its sequence similarity to IL-17A. Analyses of molecular bases for differential physiological effects between IL-17A and IL-25 are desired, although such an issue is often difficult to study due to highly complex, context-dependent, non-specific, and degenerate features of intracellular signal transduction. It is likely that there are many more factors regulating this subtle balance; for instance, T cell expression of E-FABP promotes Th17 differentiation, while counterregulating development of Tregs [241]. E-FABP-deficient CD4+ T cells display enhanced PPARγ expression and activity. This is an example of the intricate regulation of Treg/Th17 balance via lipid metabolism. It could be that FABPs act to sequester PPAR ligands in the cytoplasm, hence inhibiting nuclear entry and PPAR activity [241]. A deep understanding of lipid metabolism may enable fine-tuning of the balance between Treg and Th17 cells, and among Th17 subsets.
From a clinical perspective, the availability of drugs that explicitly act on specific cells or accumulate in local tissues/organs may become important, given that systemic drug administration is generally apt to lead to adverse events. It would be interesting to envisage that, in future, gene transduction could enable controllable accumulation of regulatory cells to a particular target organ so that the local Treg/Th17 balance can be modulated, without changing Th17 levels. As therapeutic approaches using aptamers exemplify, future therapeutics will be more directed toward local and cell type-specific interventions. To facilitate such organ/cell-specific approaches, understanding of the trafficking of effector T cells is important. CCR6 helps to recruit Th17 cells to inflamed tissues/organs in response to its ligand CCL20 [e.g., 334]. Genetic studies have demonstrated an association between susceptibility to RA and polymorphism of the gene for CCR6, the important chemokine receptor of Th17 [335]. Association of CCR6 with lupus nephritis has also been reported [336]. Koga et al. assessed the effects of suppression of the CCR6/CCL20 axis and showed reduced kidney CCR6/CCL20 expression and serum IL-17 levels, as well as improved clinical and pathologic outcomes in a lupus nephritis-like kidney disease mouse model [337]. While these findings are informative, the migratory properties of effector T cells are poorly understood in general. For instance, Elhofy et al. showed that CCR6 is largely dispensable for EAE pathogenesis [338]; CCR6-deficient mice developed a significantly more severe chronic EAE compared with wild-type immunized animals. It should also be borne in mind that chemokines and their receptors have not been identified exhaustively. Kara et al. showed IL-23-dependent switches from CCR6 to CCR2 usage during Th17 cell development (Figure 1). This switch gives rise to CCR6-CCR2+ Th17 cells that represent an advanced differentiated state producing GM-CSF/IFN-γ and therefore has a very high proinflammatory potency [20]. It would be interesting to attempt administration of Tregs transfected with genes for CCR2 in autoimmune disease model mice.
Fine-tuning of Treg/Th17, and of the subsets of Th17 cells, appears important in the intestine, in particular. While clinical trials using IL-17A inhibitors for psoriasis, ankylosing spondylitis, and RA showed promising results, the trials on Crohn’s disease instead showed increased disease activity and adverse events [339,340]. Inhibition of IL-17 activity should lead to susceptibility to infection. IL-17 and IL-22 produced by Th17 are considered to be important for epithelial cell production of β-defensin that has antifungal activity, and inhibition of this loop may lead to increases in fungi, leading to an enhanced innate immunity response in intestinal mucosa [341].
Studies on the roles of antigen recognition by TCR in autoimmune diseases have been relatively limited, but could be important. Induction of antigen-specific tolerance using chemically modified antigen peptides seems to be an interesting approach. An obvious challenge in clinics, however, would be that the cDNA sequences of TCRs and immunoglobulins from patients tell us little about the antigen peptides key to the patient. In Ito et al.’s study [100], mice expressing a single arthritogenic TCR were used for identification of peptide antigen, and this identification required auto-antibody/B cell analysis. This is elegant but still very intensive work. It would be nonetheless interesting to envisage that this approach could generate a panel of peptides from autoimmune arthritis in mice, and this information may facilitate, in combination with appropriate conjugation methods, induction of anergy of Th1 and Th17 cells in patients with RA [103].
References
- Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG (2013) The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol 34: 174-181. [Crossref]
- Koenders MI, van den Berg WB (2015) Novel therapeutic targets in rheumatoid arthritis. Trends Pharmacol Sci 36: 189-195. [Crossref]
- Kim BS, Park YJ, Chung Y (2016) Targeting IL-17 in autoimmunity and inflammation. Arch Pharm Res. [Crossref]
- Kim GW, Lee NR, Pi RH, Lim YS, Lee YM, et al. (2015) IL-6 inhibitors for treatment of rheumatoid arthritis: past, present, and future. Arch Pharm Res 38: 575-584. [Crossref]
- Yoshida H, Hashizume M, Mihara M (2011) IL-6 blockade preferentially inhibits Th17 differentiation in collagen-induced arthritis. Rheumatol Int 31: 127-131. [Crossref]
- Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, et al. (1996) T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med 183: 2593-2603. [Crossref]
- Beringer A, Noack M, Miossec P (2016) IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol Med 22: 230-241. [Crossref]
- Waisman A, Hauptmann J, Regen T (2015) The role of IL-17 in CNS diseases. Acta Neuropathol 129: 625-637. [Crossref]
- Taleb S, Tedgui A, Mallat Z (2015) IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb Vasc Biol 35: 258-264. [Crossref]
- Hou L, Wang T, Sun J (2015) T cells in infection and autoimmunity. Int Immunopharmacol 28: 887-891. [Crossref]
- Sutton CE, Mielke LA, Mills KH (2012) IL-17-producing γδ T cells and innate lymphoid cells. Eur J Immunol 42: 2221-2231. [Crossref]
- Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27: 485-517. [Crossref]
- Gaffen SL, Hernández-Santos N, Peterson AC (2011) IL-17 signaling in host defense against Candida albicans. Immunol Res 50: 181-187. [Crossref]
- Siakavellas SI, Bamias G (2012) Role of the IL-23/IL-17 axis in Crohn's disease. Discov Med 14: 253-262. [Crossref]
- Sun D, Liang D, Kaplan HJ, Shao H (2015) The role of Th17-associated cytokines in the pathogenesis of experimental autoimmune uveitis (EAU). Cytokine 74: 76-80. [Crossref]
- Kugyelka R, Kohl Z, Olasz K, Mikecz K, Rauch TA, et al. (2016) Enigma of IL-17 and Th17 Cells in Rheumatoid Arthritis and in Autoimmune Animal Models of Arthritis. Mediators Inflamm 2016: 6145810. [Crossref]
- Dos Passos GR, Sato DK, Becker J, Fujihara K (2016) Th17 Cells Pathways in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders: Pathophysiological and Therapeutic Implications. Mediators Inflamm 2016: 5314541. [Crossref]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, et al. (2008) T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28: 29-39. [Crossref]
- Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM (2008) Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J Immunol 180: 214-221. [Crossref]
- Kara EE, McKenzie DR, Bastow CR, Gregor CE, Fenix KA, et al. (2015) CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat Commun 6: 8644. [Crossref]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, et al. (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8: 967-974. [Crossref]
- Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14: 585-600. [Crossref]
- Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, et al. (2011) RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560-567. [Crossref]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, et al. (2009) The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol 10: 314-324. [Crossref]
- Haines CJ, Chen Y, Blumenschein WM, Jain R, Chang C, et al. (2013) Autoimmune memory T helper 17 cell function and expansion are dependent on interleukin-23. Cell Rep 3: 1378-1388. [Crossref]
- Veldhoen M, Hocking RJ, Flavell RA, Stockinger B (2006) Signals msediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol 7: 1151-1156. [Crossref]
- Speck S, Lim J, Shelake S, Matka M, Stoddard J, et al. (2014) TGF-β signaling initiated in dendritic cells instructs suppressive effects on Th17 differentiation at the site of neuroinflammation. PLoS One 9: e102390. [Crossref]
- Dudakov JA, Hanash AM, van den Brink MR (2015) Interleukin-22: immunobiology and pathology. Annu Rev Immunol 33: 747-785. [Crossref]
- Rankin LC, Girard-Madoux MJ, Seillet C, et al. (2016) Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol 17: 179-186. [Crossref]
- Becher B, Segal BM (2011) T(H)17 cytokines in autoimmune neuro-inflammation. Curr Opin Immunol 23: 707-712. [Crossref]
- Luchtman DW, Ellwardt E, Larochelle C, Zipp F (2014) IL-17 and related cytokines involved in the pathology and immunotherapy of multiple sclerosis: Current and future developments. Cytokine Growth Factor Rev 25: 403-413. [Crossref]
- Roeleveld DM, Koenders MI (2015) The role of the Th17 cytokines IL-17 and IL-22 in Rheumatoid Arthritis pathogenesis and developments in cytokine immunotherapy. Cytokine 74: 101-107. [Crossref]
- Sallusto F, Zielinski CE, Lanzavecchia A (2012) Human Th17 subsets. Eur J Immunol 42: 2215-2220. [Crossref]
- Hao JQ (2014) Targeting interleukin-22 in psoriasis. Inflammation 37: 94-99. [Crossref]
- Horiuchi S, Onodera A, Hosokawa H, Watanabe Y, Tanaka T, et al. (2011) Genome-wide analysis reveals unique regulation of transcription of Th2-specific genes by GATA3. J Immunol 186: 6378-6389. [Crossref]
- Kurebayashi Y, Nagai S, Ikejiri A, Koyasu S (2013) Recent advances in understanding the molecular mechanisms of the development and function of Th17 cells. Genes Cells 18: 247-265. [Crossref]
- Nagai S, Kurebayashi Y, Koyasu S (2013) Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci 1280: 30-34. [Crossref]
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, et al. (2011) The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12: 295-303. [Crossref]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, et al. (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317: 256-260. [Crossref]
- Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, et al. (2008) Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111: 1013-1020. [Crossref]
- Huber S, Gagliani N, Esplugues E, O'Connor W Jr, Huber FJ, et al. (2011) Th17 cells express interleukin-10 receptor and are controlled by Foxp3- and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34: 554-655. [Crossref]
- Heo YJ, Joo YB, Oh HJ, Park MK, Heo YM, et al. (2010) IL-10 suppresses Th17 cells and promotes regulatory T cells in the CD4+ T cell population of rheumatoid arthritis patients. Immunol Lett 127: 150-156. [Crossref]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, et al. (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133-1141. [Crossref]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, et al. (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6: 1123-1132. [Crossref]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, et al. (2008) TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453: 236-240. [Crossref]
- Huber M, Lohoff M (2014) IRF4 at the crossroads of effector T-cell fate decision. Eur J Immunol 44: 1886-1895. [Crossref]
- Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, et al. (2009) The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med 206: 2079-2089. [Crossref]
- Cui G, Qin X, Wu L, Zhang Y, Sheng X, et al. (2011) Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest 121: 658-670. [Crossref]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, et al. (2007) Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26: 371-381. [Crossref]
- Hermann-Kleiter N, Gruber T, Lutz-Nicoladoni C, Thuille N, Fresser F, et al. (2008) The nuclear orphan receptor NR2F6 suppresses lymphocyte activation and T helper 17-dependent autoimmunity. Immunity 29: 205-216. [Crossref]
- Ichiyama K, Hashimoto M, Sekiya T, Nakagawa R, Wakabayashi Y, et al. (2009) Gfi1 negatively regulates T(h)17 differentiation by inhibiting RORgammat activity. Int Immunol 21: 881-889. [Crossref]
- Carow B, Rottenberg ME (2014) SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol 5: 58. [Crossref]
- Cejas PJ, Walsh MC, Pearce EL, Han D, Harms GM, et al. (2010) TRAF6 inhibits Th17 differentiation and TGF-beta-mediated suppression of IL-2. Blood 115: 4750-4757. [Crossref]
- Pierau M, Engelmann S, Reinhold D, Lapp T, Schraven B, et al. (2009) Protein kinase B/Akt signals impair Th17 differentiation and support natural regulatory T cell function and induced regulatory T cell formation. J Immunol 183: 6124-6134. [Crossref]
- Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho IC (2007) Ets-1 is a negative regulator of Th17 differentiation. J Exp Med 204: 2825-2835. [Crossref]
- Na SY, Park MJ, Park S, Lee ES (2016) MicroRNA-155 regulates the Th17 immune response by targeting Ets-1 in Behçet's disease. Clin Exp Rheumatol 34 Suppl 102: 56-63. [Crossref]
- Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, et al. (2005) IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol 175: 788-795. [Crossref]
- Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, et al. (2007) IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol 8: 369-377. [Crossref]
- Wüthrich M, Gern B, Hung CY, Ersland K, Rocco N, et al. (2011) Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin Invest 121: 554-568. [Crossref]
- Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, et al. (2011) Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35: 997-1009. [Crossref]
- Warfel JM, Merkel TJ (2013) Bordetella pertussis infection induces a mucosal IL-17 response and long-lived Th17 and Th1 immune memory cells in nonhuman primates. Mucosal Immunol 6: 787-796. [Crossref]
- Papp K, Thaçi D, Reich K, Riedl E, Langley RG, et al. (2015) Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol 173: 930-939. [Crossref]
- Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, et al. (2015) IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol 16: 286-295. [Crossref]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24: 179-189. [Crossref]
- LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, et al. (2007) Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8: 630-638. [Crossref]
- Xu L, Kitani A, Fuss I, Strober W (2007) Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol 178: 6725-6729. [Crossref]
- Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, et al. (2011) CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34: 422-434. [Crossref]
- Chen Y, Haines CJ, Gutcher I, Hochweller K, Blumenschein WM, et al. (2011) Foxp3(+) regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity 34: 409-421. [Crossref]
- Boniface K, Blom B, Liu YJ, de Waal Malefyt R (2008) From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev 226: 132-146. [Crossref]
- Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, et al. (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20: 62-68. [Crossref]
- Andersson KM, Cavallini NF, Hu D, Brisslert M, Cialic R, et al. (2015) Pathogenic transdifferentiation of Th17 cells contribute to perpetuation of rheumatoid arthritis during Anti-TNF Treatment. Mol Med 21: 536-543. [Crossref]
- Ueno A, Jijon H, Chan R, Ford K, Hirota C, et al. (2013) increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm Bowel Dis 19: 2522-2534. [Crossref]
- Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, et al. (2015) Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523: 221-225. [Crossref]
- Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, et al. (2015) Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163: 1400-1412. [Crossref]
- Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, et al. (2015) CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163: 1413-1427. [Crossref]
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, et al. (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467: 967-971. [Crossref]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, et al. (2012) Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13: 991-999. [Crossref]
- Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, et al. (2012) Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484: 514-518. [Crossref]
- Soroosh P, Wu J, Xue X, Song J, Sutton SW, et al. (2014) Oxysterols are agonist ligands of RORγt and drive Th17 cell differentiation. Proc Natl Acad Sci U S A 111: 12163-12168. [Crossref]
- Setoguchi R, Hori S, Takahashi T, Sakaguchi S (2005) Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 201: 723-735. [Crossref]
- Kosmaczewska A, Ciszak L, Swierkot J, Szteblich A, Kosciow K, et al. (2015) Exogenous IL-2 controls the balance in Th1, Th17, and Treg cell distribution in patients with progressive rheumatoid arthritis treated with TNF-alpha inhibitors. Inflammation 38: 765-774. [Crossref]
- Rosenzwajg M, Churlaud G, Mallone R, Six A, Dérian N, et al. (2015) Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 58: 48-58. [Crossref]
- Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, et al. (2009) In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med 206: 751-760. [Crossref]
- Wang H, Hou L, Kwak D, Fassett J, Xu X, et al. (2016) Increasing Regulatory T Cells with Interleukin-2 and Interleukin-2 Antibody Complexes Attenuates Lung Inflammation and Heart Failure Progression. Hypertension 68: 114-122. [Crossref]
- Dittmer J (2003) The biology of the Ets1 proto-oncogene. Mol Cancer 2: 29. [Crossref]
- Tsao HW, Tai TS, Tseng W, Chang HH, Grenningloh R, et al. (2013) Ets-1 facilitates nuclear entry of NFAT proteins and their recruitment to the IL-2 promoter. Proc Natl Acad Sci U S A 110: 15776-15781. [Crossref]
- Zelante T, Wong AY, Ping TJ, Chen J, Sumatoh HR, et al. (2015) CD103(+) dendritic cells control Th17 cell function in the lung. Cell Rep 12: 1789-1801. [Crossref]
- Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, et al. (1994) Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med 180: 1961-1966. [Crossref]
- Sheikh SZ, Hegazi RA, Kobayashi T, Onyiah JC, Russo SM, et al. (2011) An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol 186: 5506-5513. [Crossref]
- Mueller R, Krahl T, Sarvetnick N (1996) Pancreatic expression of interleukin-4 abrogates insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 184: 1093-1099. [Crossref]
- Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, et al. (2003) Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med 9: 40-46. [Crossref]
- Guenova E, Skabytska Y, Hoetzenecker W, Weindl G, Sauer K, et al. (2015) IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U S A 112: 2163-2168. [Crossref]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, et al. (2009) CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326: 986-991. [Crossref]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, et al. (2009) The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10: 595-602. [Crossref]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, et al. (2009) Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458: 351-356. [Crossref]
- Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, et al. (2011) GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest 121: 4503-4515. [Crossref]
- Nosko A, Kluger MA, Diefenhardt P, Melderis S, Wegscheid C, et al. (2016) T-Bet enhances regulatory T cell fitness and directs control of Th1 responses in crescentic GN. J Am Soc Nephrol. [Crossref]
- Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, et al. (2007) Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med 204: 1335-1347. [Crossref]
- Kluger MA, Melderis S, Nosko A, Goerke B, Luig M, et al. (2016) Treg17 cells are programmed by Stat3 to suppress Th17 responses in systemic lupus. Kidney Int 89: 158-166. [Crossref]
- Ito Y, Hashimoto M, Hirota K, Ohkura N, Morikawa H, et al. (2014) Detection of T cell responses to a ubiquitous cellular protein in autoimmune disease. Science 346: 363-368. [Crossref]
- Apostolopoulos V, Thalhammer T, Tzakos AG, Stojanovska L (2013) Targeting antigens to dendritic cell receptors for vaccine development. J Drug Deliv 2013: 869718. [Crossref]
- Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, et al. (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 194: 769-779. [Crossref]
- Tseveleki V, Tselios T, Kanistras I, Koutsoni O, Karamita M, et al. (2015) Mannan-conjugated myelin peptides prime non-pathogenic Th1 and Th17 cells and ameliorate experimental autoimmune encephalomyelitis. Exp Neurol 267: 254-267. [Crossref]
- Jenkins MK, Taylor PS, Norton SD, Urdahl KB (1991) CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol 147: 2461-2466. [Crossref]
- Rosenblum MD, Gratz IK, Paw JS, Abbas AK (2012) Treating human autoimmunity: current practice and future prospects. Sci Transl Med 4: 125sr1. [Crossref]
- Perrin PJ, Scott D, Davis TA, Gray GS, Doggett MJ, et al. (1996) Opposing effects of CTLA4-Ig and anti-CD80 (B7-1) plus anti-CD86 (B7-2) on experimental allergic encephalomyelitis. J Neuroimmunol 65: 31-39. [Crossref]
- Amezcua-Guerra LM, Hernández-Martínez B, Pineda C, Bojalil R (2006) Ulcerative colitis during CTLA-4Ig therapy in a patient with rheumatoid arthritis. Gut 55: 1059-1060. [Crossref]
- Bouguermouh S, Fortin G, Baba N, Rubio M, Sarfati M (2009) CD28 co-stimulation down regulates Th17 development. PLoS One 4: e5087. [Crossref]
- Ying H, Yang L, Qiao G, Li Z, Zhang L, et al. (2010) Cutting edge: CTLA-4--B7 interaction suppresses Th17 cell differentiation. J Immunol 185: 1375-1378. [Crossref]
- Watanabe N, Kaminum2021 Copyright OAT. All rights reserved Treg cells augment the Th17-mediated intestinal inflammatory response in a CTLA4-dependent manner. PLoS One 11: e0150244. [Crossref]
- Lanz TV, Ding Z, Ho PP, Luo J, Agrawal AN, et al. (2010) Angiotensin II sustains brain inflammation in mice via TGF-beta. J Clin Invest 120: 2782-2794. [Crossref]
- Racke MK, Dhib-Jalbut S, Cannella B, Albert PS, Raine CS, et al. (1991) Prevention and treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor-beta 1. J Immunol 146: 3012-3017. [Crossref]
- Johns LD, Sriram S (1993) Experimental allergic encephalomyelitis: neutralizing antibody to TGF beta 1 enhances the clinical severity of the disease. J Neuroimmunol 47: 1-7. [Crossref]
- Guo Y, Brown C, Ortiz C, Noelle RJ (2015) Leukocyte homing, fate, and function are controlled by retinoic acid. Physiol Rev 95: 125-148. [Crossref]
- Hall JA, Grainger JR, Spencer SP, Belkaid Y (2011) The role of retinoic acid in tolerance and immunity. Immunity 35: 13-22. [Crossref]
- Raverdeau M, Mills KH (2014) Modulation of T cell and innate immune responses by retinoic Acid. J Immunol 192: 2953-2958. [Crossref]
- Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B (2007) Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol 8: 1086-1094.
- Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciumè G, et al. (2012) TGF-ß and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol 13: 587-595. [Crossref]
- Pino-Lagos K, Guo Y, Brown C, Alexander MP, Elgueta R, et al. (2011) A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. J Exp Med 208: 1767-1775. [Crossref]
- Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, et al. (2010) Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity 33: 313-325. [Crossref]
- Iwata M1, Hirakiyama A, Eshima Y, Kagechika H, Kato C, et al. (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21: 527-538. [Crossref]
- Ruane D, Brane L, Reis BS, Cheong C, Poles J, et al. (2013) Lung dendritic cells induce migration of protective T cells to the gastrointestinal tract. J Exp Med 210: 1871-1888. [Crossref]
- Molenaar R, Greuter M, van der Marel AP, Roozendaal R, Martin SF, et al. (2009) Lymph node stromal cells support dendritic cell-induced gut-homing of T cells. J Immunol 183: 6395-6402. [Crossref]
- Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, et al. (2008) Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med 205: 2139-2149. [Crossref]
- Cerovic V, Bain CC, Mowat AM, Milling SW (2014) Intestinal macrophages and dendritic cells: what's the difference? Trends Immunol 35: 270-277. [Crossref]
- Welty NE, Staley C, Ghilardi N, Sadowsky MJ, Igyártó BZ, et al. (2013) Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J Exp Med 210: 2011-2024. [Crossref]
- Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, et al. (2013) Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J Exp Med 210: 1961-1976. [Crossref]
- Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, et al. (2009) Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med 15: 401-409. [Crossref]
- Yokota A, Takeuchi H, Maeda N, Ohoka Y, Kato C, et al. (2009) GM-CSF and IL-4 synergistically trigger dendritic cells to acquire retinoic acid-producing capacity. Int Immunol 21: 361-377. [Crossref]
- Elgueta R, Sepulveda FE, Vilches F, Vargas L, Mora JR, et al. (2008) Imprinting of CCR9 on CD4 T cells requires IL-4 signaling on mesenteric lymph node dendritic cells. J Immunol 180: 6501-6507. [Crossref]
- Wang S, Villablanca EJ, De Calisto J, Gomes DC, Nguyen DD, et al. (2011) MyD88-dependent TLR1/2 signals educate dendritic cells with gut-specific imprinting properties. J Immunol 187: 141-150. [Crossref]
- Lu L, Ma J, Li Z, Lan Q, Chen M, et al. (2011) All-trans retinoic acid promotes TGF-β-induced Tregs via histone modification but not DNA demethylation on Foxp3 gene locus. PLoS One 6: e24590. [Crossref]
- Nguyen V, Pearson K, Kim JH, Kamdar K, DePaolo RW (2015) Retinoic acid can exacerbate T cell intrinsic TLR2 activation to promote tolerance. PLoS One 10: e0118875. [Crossref]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. (2011) The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332: 974-977. [Crossref]
- Wu J, Zhang Y, Liu Q, Zhong W, Xia Z (2013) All-trans retinoic acid attenuates airway inflammation by inhibiting Th2 and Th17 response in experimental allergic asthma. BMC Immunol 14: 28. [Crossref]
- Honarvar NM, Harrirchian MH, Koohdani F, Siassi F, Jafari Rad S, et al. (2013) In vitro effect of human serum and fetal calf serum on CD4+ T cells proliferation in response to myelin oligodendrocyte glycoprotein (MOG) in correlation with RBP/TTR ratio in multiple sclerotic patients. J Mol Neurosci 50: 571-576. [Crossref]
- Saboor-Yaraghi AA, Harirchian MH, Mohammadzadeh Honarvar N, Bitarafan S, Abdolahi M, et al. (2015) The Effect of Vitamin A Supplementation on FoxP3 and TGF-ß Gene Expression in Avonex-Treated Multiple Sclerosis Patients. J Mol Neurosci 56: 608-612. [Crossref]
- Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, et al. (2010) Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329: 849-853. [Crossref]
- Germain P, Iyer J, Zechel C, Gronemeyer H (2002) Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature 415: 187-192. [Crossref]
- Chandraratna RA, Noelle RJ, Nowak EC (2016) Treatment with retinoid X receptor agonist IRX4204 ameliorates experimental autoimmune encephalomyelitis. Am J Transl Res 8: 1016-1026. [Crossref]
- Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, et al. (2015) Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 42: 499-511. [Crossref]
- Hayes CE, Hubler SL, Moore JR, Barta LE, Praska CE, et al. (2015) Vitamin D Actions on CD4(+) T Cells in Autoimmune Disease. Front Immunol 6: 100. [Crossref]
- Ascherio A, Munger KL, White R, Köchert K, Simon KC, et al. (2014) Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol 71: 306-314. [Crossref]
- Burton JM, Kimball S, Vieth R, Bar-Or A, Dosch HM, et al. (2010) A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology 74: 1852-1859. [Crossref]
- Pozuelo-Moyano B, Benito-León J, Mitchell AJ, Hernández-Gallego J (2013) A systematic review of randomized, double-blind, placebo-controlled trials examining the clinical efficacy of vitamin D in multiple sclerosis. Neuroepidemiology 40: 147-153. [Crossref]
- Smolders J, Peelen E, Thewissen M, Menheere P, Tervaert JW, et al. (2009) The relevance of vitamin D receptor gene polymorphisms for vitamin D research in multiple sclerosis. Autoimmun Rev 8: 621-626. [Crossref]
- Berlanga-Taylor AJ, Disanto G, Ebers GC, Ramagopalan SV (2011) Vitamin D-gene interactions in multiple sclerosis. J Neurol Sci 311: 32-36. [Crossref]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476: 214-219. [Crossref]
- Shahijanian F, Parnell GP, McKay FC, Gatt PN, Shojoei M, et al. (2014) The CYP27B1 variant associated with an increased risk of autoimmune disease is underexpressed in tolerizing dendritic cells. Hum Mol Genet 23: 1425-1434. [Crossref]
- Daniel C , Sartory NA , Zahn N , Radeke HH , Stein JM (2008) Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther 324: 23-33. [Crossref]
- Mayne CG, Spanier JA, Relland LM, Williams CB, Hayes CE (2011) 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol 41: 822-832. [Crossref]
- Bruce D, Yu S, Ooi JH, Cantorna MT (2011) Converging pathways lead to overproduction of IL-17 in the absence of vitamin D signaling. Int Immunol 23: 519-528. [Crossref]
- Tiosano D, Wildbaum G, Gepstein V, Verbitsky O, Weisman Y, et al. (2013) The role of vitamin D receptor in innate and adaptive immunity: a study in hereditary vitamin D-resistant rickets patients. J Clin Endocrinol Metab 98: 1685-1693. [Crossref]
- Subramaniam N, Leong GM, Cock TA, Flanagan JL, Fong C, et al. (2001) Cross-talk between 1,25-dihydroxyvitamin D3 and transforming growth factor-beta signaling requires binding of VDR and Smad3 proteins to their cognate DNA recognition elements. J Biol Chem 276: 15741-15746.
- Nanduri R, Mahajan S, Bhagyaraj E, Sethi K, Kalra R, et al. (2015) The active form of vitamin d transcriptionally represses Smad7 signaling and activates extracellular signal-regulated kinase (ERK) to inhibit the differentiation of a inflammatory T helper cell subset and suppress experimental autoimmune encephalomyelitis. J Biol Chem 290: 12222-12236. [Crossref]
- Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, et al. (1997) Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389: 631-635. [Crossref]
- Milliken SV, Wassall H, Lewis BJ, Logie J, Barker RN, et al. (2012) Effects of ultraviolet light on human serum 25-hydroxyvitamin D and systemic immune function. J Allergy Clin Immunol 129: 1554-1561.
- Peters A, Fowler KD, Chalmin F, Merkler D, Kuchroo VK, et al. (2015) IL-27 Induces Th17 Differentiation in the Absence of STAT1 Signaling. J Immunol 195: 4144-4153. [Crossref]
- Neufert C, Becker C, Wirtz S, Fantini MC, Weigmann B, et al. (2007) IL-27 controls the development of inducible regulatory T cells and Th17 cells via differential effects on STAT1. Eur J Immunol 37: 1809-1816. [Crossref]
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciumè G, et al. (2015) Asymmetric action of STAT transcription factors drives transcriptional outputs and cytokine specificity. Immunity 42: 877-889. [Crossref]
- Grogan JL, Ouyang W (2012) A role for Th17 cells in the regulation of tertiary lymphoid follicles. Eur J Immunol 42: 2255-2262. [Crossref]
- Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, et al. (2011) Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 472: 486-490. [Crossref]
- Fujita-Sato S, Ito S, Isobe T, Ohyama T, Wakabayashi K, et al. (2011) Structural basis of digoxin that antagonizes RORgamma t receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production. J Biol Chem 286: 31409-31417. [Crossref]
- Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, et al. (2011) Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472: 491-494. [Crossref]
- Xiao S, Yosef N, Yang J, Wang Y, Zhou L, et al. (2014) Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 40: 477-489. [Crossref]
- Lin H, Song P, Zhao Y, Xue LJ, Liu Y, et al. (2015) Targeting Th17 cells with small molecules and small interference rna. Mediators Inflamm 2015: 290657. [Crossref]
- Xue Y, Yang Y, Su Z, Barnie PA, Zheng D, et al. (2014) Local delivery of T-bet shRNA reduces inflammation in collagen II-induced arthritis via downregulation of IFN-γ and IL-17. Mol Med Rep 9: 899-903. [Crossref]
- Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch'en IL, et al. (2010) Foxo transcription factors control regulatory T cell development and function. Immunity 33: 890-904. [Crossref]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, et al. (2012) Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491: 554-559. [Crossref]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, et al. (2013) Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496: 513-517. [Crossref]
- Lainé A, Martin B, Luka M, Mir L, Auffray C, et al. (2015) Foxo1 is a T cell-intrinsic inhibitor of the RORγt-Th17 program. J Immunol 195: 1791-1803. [Crossref]
- Devergne O, Birkenbach M, Kieff E (1997) Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc Natl Acad Sci U S A 94: 12041-12046. [Crossref]
- Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, et al. (2010) IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol 11: 1093-1101. [Crossref]
- Egwuagu CE, Yu CR, Sun L, Wang R (2015) Interleukin 35: Critical regulator of immunity and lymphocyte-mediated diseases. Cytokine Growth Factor Rev 26: 587-593. [Crossref]
- Choi J, Leung PS, Bowlus C, Gershwin ME (2015) IL-35 and Autoimmunity: a Comprehensive Perspective. Clin Rev Allergy Immunol 49: 327-332. [Crossref]
- Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, et al. (2007) IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol 37: 3021-3029. [Crossref]
- Wirtz S, Billmeier U, Mchedlidze T, Blumberg RS, Neurath MF (2011) Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 141: 1875-1886. [Crossref]
- Bettini M, Castellaw AH, Lennon GP, Burton AR, Vignali DA (2012) Prevention of autoimmune diabetes by ectopic pancreatic β-cell expression of interleukin-35. Diabetes 61: 1519-1526. [Crossref]
- Kochetkova I, Golden S, Holderness K, Callis G, Pascual DW (2010) IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J Immunol 184: 7144-7153. [Crossref]
- Thiolat A, Denys A, Petit M, Biton J, Lemeiter D, et al. (2014) Interleukin-35 gene therapy exacerbates experimental rheumatoid arthritis in mice. Cytokine 69: 87-93. [Crossref]
- Filková M, Vernerová Z, Hulejová H, Prajzlerová K, Veigl D, et al. (2015) Pro-inflammatory effects of interleukin-35 in rheumatoid arthritis. Cytokine 73: 36-43. [Crossref]
- Cao J, Xu F, Lin S, Tao X, Xiang Y, et al. (2015) IL-35 is elevated in clinical and experimental sepsis and mediates inflammation. Clin Immunol 161: 89-95. [Crossref]
- Varikuti S, Oghumu S, Natarajan G, Kimble J, Sperling RH, et al. (2016) STAT4 is required for the generation of Th1 and Th2, but not Th17 immune responses during monophosphoryl lipid A adjuvant activity. Int Immunol pii: dxw038. [Crossref]
- McWilliams IL, Rajbhandari R, Nozell S, Benveniste E, Harrington LE (2015) STAT4 controls GM-CSF production by both Th1 and Th17 cells during EAE. J Neuroinflammation 12: 128. [Crossref]
- Dulek DE, Newcomb DC, Toki S, Goliniewska K, Cephus J, et al. (2014) STAT4 deficiency fails to induce lung Th2 or Th17 immunity following primary or secondary respiratory syncytial virus (RSV) challenge but enhances the lung RSV-specific CD8+ T cell immune response to secondary challenge. J Virol 88: 9655-9672. [Crossref]
- Ma J, Wang R, Fang X, Sun Z (2012) β-catenin/TCF-1 pathway in T cell development and differentiation. J Neuroimmune Pharmacol 7: 750-762. [Crossref]
- Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, et al. (2010) Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol 8: e1000296. [Crossref]
- Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ (2008) Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med 14: 162-169. [Crossref]
- Schilham MW, Wilson A, Moerer P, Benaissa-Trouw BJ, Cumano A, et al. (1998) Critical involvement of Tcf-1 in expansion of thymocytes. J Immunol 161: 3984-3991. [Crossref]
- Ma J, Wang R, Fang X, Ding Y, Sun Z (2011) Critical role of TCF-1 in repression of the IL-17 gene. PLoS One 6: e24768. [Crossref]
- Zhou L (2016) AHR function in lymphocytes: emerging concepts. Trends Immunol 37: 17-31. [Crossref]
- Liu HP, Cao AT, Feng T, Li Q, Zhang W, et al. (2015) TGF-β converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol 45: 1010-1018. [Crossref]
- Ye H, Zhang J, Wang J, Gao Y, Du Y, et al. (2015) CD4 T-cell transcriptome analysis reveals aberrant regulation of STAT3 and Wnt signaling pathways in rheumatoid arthritis: evidence from a case-control study. Arthritis Res Ther 17: 76. [Crossref]
- Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez-Rios J (2008) Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci 121: 737-746. [Crossref]
- Lee YS, Lee KA, Yoon HB, Yoo SA, Park YW, et al. (2012) The Wnt inhibitor secreted Frizzled-Related Protein 1 (sFRP1) promotes human Th17 differentiation. Eur J Immunol 42: 2564-2573. [Crossref]
- Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, et al. (2015) TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 125: 4053-4062. [Crossref]
- Joller N, Lozano E, Burkett PR, Patel B, Xiao S, et al. (2014) Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40: 569-581. [Crossref]
- Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, et al. (2011) Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol 41: 902-915. [Crossref]
- Zhao W, Dong Y, Wu C, Ma Y, Jin Y, et al. (2016) TIGIT overexpression diminishes the function of CD4 T cells and ameliorates the severity of rheumatoid arthritis in mouse models. Exp Cell Res 340: 132-138. [Crossref]
- Wang J, Vuitton DA, Müller N, Hemphill A, Spiliotis M, et al. (2015) Deletion of fibrinogen-like protein 2 (FGL-2), a novel CD4+ CD25+ Treg effector molecule, leads to improved control of echinococcus multilocularis infection in mice. PLoS Negl Trop Dis 9: e0003755. [Crossref]
- Fanzo JC, Yang W, Jang SY, Gupta S, Chen Q, et al. (2006) Loss of IRF-4-binding protein leads to the spontaneous development of systemic autoimmunity. J Clin Invest 116: 703-714. [Crossref]
- Chen Q, Yang W, Gupta S, Biswas P, Smith P, et al. (2008) IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity 29: 899-911. [Crossref]
- Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, et al. (2010) Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest 120: 3280-3295. [Crossref]
- Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, et al. (2014) Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A 111: 16814-16819. [Crossref]
- Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, et al. (2016) Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 127: 2144-2154. [Crossref]
- Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, et al. (2009) The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460: 405-409. [Crossref]
- Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, et al. (2012) A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338: 975-980. [Crossref]
- Miao T, Raymond M, Bhullar P, Ghaffari E, Symonds AL, et al. (2013) Early growth response gene-2 controls IL-17 expression and Th17 differentiation by negatively regulating Batf. J Immunol 190: 58-65. [Crossref]
- Zhu B, Symonds AL, Martin JE, Kioussis D, Wraith DC, et al. (2008) Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med 205: 2295-2307. [Crossref]
- Ouyang X, Zhang R, Yang J, Li Q, Qin L, et al. (2011) Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat Commun 2: 314. [Crossref]
- Waickman AT, Powell JD (2012) mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev 249: 43-58. [Crossref]
- Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, et al. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153: 1239-1251. [Crossref]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, et al. (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64: 985-993. [Crossref]
- Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, et al. (2013) Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14: 500–508. [Crossref]
- Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, et al. (2014) Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40: 692-705. [Crossref]
- Jewell JL, Russell RC, Guan KL (2013) Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 14: 133-139. [Crossref]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, et al. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290-303. [Crossref]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, et al. (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171-183. [Crossref]
- Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, et al. (2007) The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev 21: 1037-1049. [Crossref]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, et al. (2011) HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208: 1367-1376. [Crossref]
- Kastirr I, Crosti M, Maglie S, Paroni M, Steckel B, et al. (2015) Signal strength and metabolic requirements control cytokine-induced Th17 differentiation of uncommitted human T cells. J Immunol 195: 3617-3627. [Crossref]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, et al. (2014) The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20: 61-72. [Crossref]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, et al. (1998) Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18: 38-43. [Crossref]
- Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577-590. [Crossref]
- Hardie DG (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8: 774-785. [Crossref]
- MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, et al. (2011) The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol 187: 4187-4198. [Crossref]
- Wang R, Solt LA (2016) Metabolism of murine TH 17 cells: Impact on cell fate and function. Eur J Immunol 46: 807-816. [Crossref]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, et al. (2011) Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186: 3299-3303. [Crossref]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, et al. (2013) Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19: 1649-1654.
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, et al. (2014) De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20: 1327-1333.
- Kim JH, Song J, Park KW (2015) The multifaceted factor peroxisome proliferator-activated receptorγ (PPARγ) in metabolism, immunity, and cancer. Arch Pharm Res 38: 302-312. [Crossref]
- da Rocha Junior LF, Dantas AT, Duarte AL, de Melo Rego MJ, Pitta Ida R, et al. (2013) PPARγ agonists in adaptive immunity: What do immune disorders and their models have to tell us? PPAR Res 2013: 519724. [Crossref]
- Mottis A, Mouchiroud L, Auwerx J (2013) Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 27: 819-835. [Crossref]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, et al. (2007) Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell 25: 57-70. [Crossref]
- Zhu R, Ou Z, Ruan X, Gong J (2012) Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol Med Rep 5: 895-900. [Crossref]
- Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, et al. (2004) LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119: 299-309. [Crossref]
- Walcher D, Kümmel A, Kehrle B, Bach H, Grüb M, et al. (2006) LXR activation reduces proinflammatory cytokine expression in human CD4-positive lymphocytes. Arterioscler Thromb Vasc Biol 26: 1022-1028. [Crossref]
- Chen S, Sorrentino R, Shimada K, Bulut Y, Doherty TM, et al. (2008) Chlamydia pneumoniae-induced foam cell formation requires MyD88-dependent and -independent signaling and is reciprocally modulated by liver X receptor activation. J Immunol 181: 7186-7193.
- Fontaine C, Rigamonti E, Nohara A, Gervois P, Teissier E, Fruchart JC, et al. (2007) Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ Res 101: 40-49.
- Korf H, Vander Beken S, Romano M, Steffensen KR, Stijlemans B, et al. (2009) Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J Clin Invest 119: 1626-1637.
- Li B, Reynolds JM, Stout RD, Bernlohr DA, Suttles J (2009) Regulation of Th17 differentiation by epidermal fatty acid-binding protein. J Immunol 182: 7625-7633. [Crossref]
- Ayroldi E, Macchiarulo A, Riccardi C (2014) Targeting glucocorticoid side effects: selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? A perspective. FASEB J 28: 5055-5070. [Crossref]
- Cheng Q, Morand E, Yang YH (2014) Development of novel treatment strategies for inflammatory diseases-similarities and divergence between glucocorticoids and GILZ. Front Pharmacol 5: 169.
- Hoppstädter J, Kiemer AK (2015) Glucocorticoid-induced leucine zipper (GILZ) in immuno suppression: master regulator or bystander? Oncotarget 6: 38446-38457. [Crossref]
- Jones SA, Perera DN, Fan H, Russ BE, Harris J, et al. (2015) GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J Autoimmun 61: 73-80. [Crossref]
- Nishizawa K, Seki R (2016) Mechanisms of immunosuppression by mesenchymal stromal cells: a review with a focus on molecules. Biomed Res Clin Prac 1: 116.
- Luz-Crawford P, Tejedor G, Mausset-Bonnefont AL, Beaulieu E, Morand EF, et al. (2015) Glucocorticoid-induced leucine zipper governs the therapeutic potential of mesenchymal stem cells by inducing a switch from pathogenic to regulatory Th17 cells in a mouse model of collagen-induced arthritis. Arthritis Rheumatol 67: 1514-1524. [Crossref]
- Yoshida H, Hunter CA (2015) The immunobiology of interleukin-27. Annu Rev Immunol 33: 417-443. [Crossref]
- Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, et al. (2011) IL-27 mediates the response to IFN-ß therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun 25: 1170-1181. [Crossref]
- Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK (2009) Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 183: 7169-7177. [Crossref]
- Fitzgerald DC, Fonseca-Kelly Z, Cullimore ML, Safabakhsh P, Saris CJ, et al. (2013) Independent and interdependent immunoregulatory effects of IL-27, IFN-ß, and IL-10 in the suppression of human Th17 cells and murine experimental autoimmune encephalomyelitis. J Immunol 190: 3225-3234.
- Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, et al. (2007) Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol 8: 1372-1379. [Crossref]
- Tang SC, Fan XH, Pan QM, Sun QS, Liu Y (2015) Decreased expression of IL-27 and its correlation with Th1 and Th17 cells in progressive multiple sclerosis. J Neurol Sci 348: 174-180. [Crossref]
- Naderi S, Hejazi Z, Shajarian M, Alsahebfosoul F, Etemadifar M, et al. (2016) IL-27 plasma level in relapsing remitting multiple sclerosis subjects: The double-faced cytokine. J Immunoassay Immunochem 37: 659-670. [Crossref]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119-124. [Crossref]
- Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, et al. (2013) TH17 cell differentiation is regulated by the circadian clock. Science 342: 727-730. [Crossref]
- Duez H, van der Veen JN, Duhem C, Pourcet B, Touvier T, et al. (2008) Regulation of bile acid synthesis by the nuclear receptor Rev-erbalpha. Gastroenterology 135: 689-698. [Crossref]
- Farez MF, Mascanfroni ID, Méndez-Huergo SP, Yeste A, Murugaiyan G, et al. (2015) Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell 162: 1338-1352. [Crossref]
- Jin Y, de Pedro-Cuesta J, Söderström M, Stawiarz L, Link H (2000) Seasonal patterns in optic neuritis and multiple sclerosis: a meta-analysis. J Neurol Sci 181: 56-64. [Crossref]
- Spelman T, Gray O, Trojano M, Petersen T, Izquierdo G, et al. (2014) Seasonal variation of relapse rate in multiple sclerosis is latitude dependent. Ann Neurol 76: 880-890. [Crossref]
- Tan AH, Lam KP (2010) Pharmacologic inhibition of MEK-ERK signaling enhances Th17 differentiation. J Immunol 184: 1849-1857. [Crossref]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, et al. (2013) Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518-522. [Crossref]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, et al. (2013) Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496: 513-517. [Crossref]
- Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, et al. (2015) Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest 125: 4212-4222. [Crossref]
- Jörg S, Kissel J, Manzel A, Kleinewietfeld M, Haghikia A, et al. (2016) High salt drives Th17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp Neurol 279: 212-222. [Crossref]
- Binger KJ, Linker RA, Muller DN, Kleinewietfeld M (2015) Sodium chloride, SGK1, and Th17 activation. Pflugers Arch 467: 543-550. [Crossref]
- Salgado E, Bes-Rastrollo M, de Irala J, Carmona L, Gómez-Reino JJ (2015) High sodium intake is associated with self-reported rheumatoid arthritis: A cross sectional and case control analysis within the SUN cohort. Medicine (Baltimore) 94: e924. [Crossref]
- Farez MF, Fiol MP, Gaitán MI, Quintana FJ, Correale J (2015) Sodium intake is associated with increased disease activity in multiple sclerosis. J Neurol Neurosurg Psychiatry 86: 26-31
- McDonald J, Graves J, Waldman A, Lotze T, Schreiner T, et al. (2016) A case-control study of dietary salt intake in pediatric-onset multiple sclerosis. Mult Scler Relat Disord 6: 87-92. [Crossref]
- Monteleone I, Marafini I, Dinallo V, Di Fusco D, Troncone E, et al. (2016) Sodium chloride-enriched diet enhanced inflammatory cytokine production and exacerbated experimental colitis in mice. J Crohns Colitis 2016. pii: jjw139. [Crossref]
- Wen W, Wan Z, Ren K, Zhou D, Gao Q, et al. (2016) Potassium supplementation inhibits IL-17A production induced by salt loading in human T lymphocytes via p38/MAPK-SGK1 pathway. Exp Mol Pathol 100: 370-377. [Crossref]
- Levite M (2016) Dopamine and T cells: dopamine receptors and potent effects on T cells, dopamine production in T cells, and abnormalities in the dopaminergic system in T cells in autoimmune, neurological and psychiatric diseases. Acta Physiol (Oxf) 216: 42-89. [Crossref]
- Prado C, Contreras F, González H, Díaz P, Elgueta D, et al. (2012) Stimulation of dopamine receptor D5 expressed on dendritic cells potentiates Th17-mediated immunity. J Immunol 188: 3062-3070. [Crossref]
- Pacheco R, Contreras F, Zouali M (2014) The dopaminergic system in autoimmune diseases. Front Immunol 5: 117. [Crossref]
- Ferreira TB, Barros PO, Teixeira B, Cassano T, Centurião N, et al. (2014) Dopamine favors expansion of glucocorticoid-resistant IL-17-producing T cells in multiple sclerosis. Brain Behav Immun 41: 182-190. [Crossref]
- Melnikov M, Belousova O, Murugin V, Pashenkov M, Boyκo A (2016) The role of dopamine in modulation of Th-17 immune response in multiple sclerosis. J Neuroimmunol 292: 97-101. [Crossref]
- Giorelli M, Livrea P, Trojano M (2005) Dopamine fails to regulate activation of peripheral blood lymphocytes from multiple sclerosis patients: effects of IFN-γ. J Interferon Cytokine Res 25: 395-406.
- Durelli L, Conti L, Clerico M, Boselli D, Contessa G, et al. (2009) T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol 65: 499-509. [Crossref]
- Nagalingam NA, Kao JY, Young VB (2011) Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm Bowel Dis 17: 917-926. [Crossref]
- Wu HJ, Wu E (2012) The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 3: 4-14. [Crossref]
- Chervonsky AV (2013) Microbiota and autoimmunity. Cold Spring Harb Perspect Biol 5: a007294. [Crossref]
- Rosser EC, Mauri C (2016) A clinical update on the significance of the gut microbiota in systemic autoimmunity. J Autoimmun 74: 85-93. [Crossref]
- de Paiva CS, Jones DB, Stern ME, Bian F, Moore QL, et al. (2016) Altered mucosal microbiome diversity and disease severity in Sjögren syndrome. Sci Rep 6: 23561. [Crossref]
- Shimizu J, Suzuki N (2016) Enhanced Th17 responses with intestinal dysbiosis in human allergic, inflammatory, and autoimmune diseases. Biomed Res Clin Prac 1: 113.
- Omenetti S, Pizarro TT (2015) The Treg/Th17 Axis: A dynamic balance regulated by the gut microbiome. Front Immunol 6: 639. [Crossref]
- Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, et al. (2016) Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer's patch T follicular helper cells. Immunity 44: 875-888. [Crossref]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, et al. (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331: 337-341. [Crossref]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, et al. (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500: 232-236. [Crossref]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, et al. (2010) Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32: 815-827. [Crossref]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, et al. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485-498. [Crossref]
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, et al. (2015) Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163: 367-380. [Crossref]
- Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, et al. (2016) Intestinal Interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity 44: 659-671. [Crossref]
- Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, et al. (2014) Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41: 152-165. [Crossref]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, et al. (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504: 446-450. [Crossref]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504: 451-455. [Crossref]
- Nishizawa K (2016) Low-grade endotoxemia, diet, and gut microbiota – an emphasis on the early events leading to dysfunction of the intestinal epithelial barrier. Biomed Res Clin Prac 1: 110.
- Licciardi PV, Karagiannis TC (2012) Regulation of immune responses by histone deacetylase inhibitors. ISRN Hematol 2012: 690901. [Crossref]
- Kroesen M, Gielen P, Brok IC, Armandari I, Hoogerbrugge PM, et al. (2014) HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 5: 6558-6572. [Crossref]
- Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, et al. (2008) Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 112: 2340-2352. [Crossref]
- de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW (2010) Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 138: 583-594. [Crossref]
- Sugimoto K, Itoh T, Takita M, Shimoda M, Chujo D, et al. (2014) Improving allogeneic islet transplantation by suppressing Th17 and enhancing Treg with histone deacetylase inhibitors. Transpl Int 27: 408-415. [Crossref]
- Knolle MD, Rana BM, McKenzie AN (2015) IL-25 as a potential therapeutic target in allergic asthma. Immunotherapy 7: 607-610. [Crossref]
- Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, et al. (2008) Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol 181: 4299-4310.
- Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, et al. (2007) IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med 204: 161-170. [Crossref]
- Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, et al. (2008) Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med 205: 2191-2198. [Crossref]
- Su J, Chen T, Ji XY, Liu C, Yadav PK, et al. (2013) IL-25 downregulates Th1/Th17 immune response in an IL-10-dependent manner in inflammatory bowel disease. Inflamm Bowel Dis 19: 720-728. [Crossref]
- Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, et al. (2008) The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol 181: 2799-2805. [Crossref]
- Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, et al. (2010) LY243982, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum 62: 929-939. [Crossref]
- Pavelka K, Chon Y, Newmark R, Lin SL, Baumgartner S, et al. (2015) A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. Rheumatol 42: 912-919. [Crossref]
- Martin DA, Churchill M, Flores-Suarez L, Cardiel MH, Wallace D, et al. (2013) A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res Ther 15: R164. [Crossref]
- Monteleone G, Pallone F, Macdonald TT (2010) Interleukin-25: a two-edged sword in the control of immune-inflammatory responses. Cytokine Growth Factor Rev 21: 471-475. [Crossref]
- Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, et al. (2014) Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 40: 989-1001. [Crossref]
- Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T (2008) Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A 105: 9721-9726. [Crossref]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, et al. (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453: 65-71. [Crossref]
- Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, et al. (2010) Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A 107: 19961Crossref]
- Stephens GL, Wang Q, Swerdlow B, Bhat G, Kolbeck R, et al. (2013) Kynurenine 3-monooxygenase mediates inhibition of Th17 differentiation via catabolism of endogenous aryl hydrocarbon receptor ligands. Eur J Immunol 43: 1727-1734. [Crossref]
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, et al. (2011) AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol 13: 144-151. [Crossref]
- Liberal R, Grant CR, Ma Y, Csizmadia E, Jiang ZG, et al. (2016) CD39 mediated regulation of Th17-cell effector function is impaired in juvenile autoimmune liver disease. J Autoimmun 72: 102-112
- Jeker LT, Marone R (2015) Targeting microRNAs for immunomodulation. Curr Opin Pharmacol 23: 25-31. [Crossref]
- Baumjohann D, Ansel KM (2013) MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol 13: 666-678. [Crossref]
- Ueno A, Ghosh A, Hung D, Li J, Jijon H (2015) Th17 plasticity and its changes associated with inflammatory bowel disease. World J Gastroenterol 21: 12283-12295. [Crossref]
- Churov AV, Oleinik EK, Knip M (2015) MicroRNAs in rheumatoid arthritis: altered expression and diagnostic potential. Autoimmun Rev 14: 1029-1037. [Crossref]
- Wu T, Chen G (2016) miRNAs participate in ms pathological processes and its therapeutic response. Mediators Inflamm 2016: 4578230. [Crossref]
- Xu XM, Zhang HJ (2016) miRNAs as new molecular insights into inflammatory bowel disease: Crucial regulators in autoimmunity and inflammation. World J Gastroenterol 22: 2206-2218. [Crossref]
- Wang H, Flach H, Onizawa M, Wei L, McManus MT, et al. (2014) Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol 15: 393-401. [Crossref]
- Naghavian R, Ghaedi K, Kiani-Esfahani A, Ganjalikhani-Hakemi M, Etemadifar M, et al. (2015) miR-141 and miR-200a, revelation of new possible players in modulation of Th17/Treg differentiation and pathogenesis of multiple sclerosis. PLoS One 10: e0124555. [Crossref]
- Perez J, Dansou B, Hervé R, Levi C, Tamouza H, et al. (2016) Calpains released by T lymphocytes cleave TLR2 to control IL-17 expression. J Immunol 196(1): 168-181. [Crossref]
- Tawfik MK (2015) Combination of coenzyme Q10 with methotrexate suppresses Freund's complete adjuvant-induced synovial inflammation with reduced hepatotoxicity in rats: Effect on oxidative stress and inflammation. Int Immunopharmacol 24:80-87. [Crossref]
- Jhun J, Lee SH, Byun JK, Jeong JH, Kim EK, et al. (2015) Coenzyme Q10 suppresses Th17 cells and osteoclast differentiation and ameliorates experimental autoimmune arthritis mice. Immunol Lett 166:92-102. [Crossref]
- Ludwinski MW, Sun J, Hilliard B, Gong S, Xue F, et al. (2009) Critical roles of Bim in T cell activation and T cell-mediated autoimmune inflammation in mice. J Clin Invest 119: 1706-1713. [Crossref]
- Iglesias M, Augustin JJ, Alvarez P, Santiuste I, Postigo J, et al. (2016) Selective impairment of TH17-differentiation and protection against autoimmune arthritis after overexpression of BCL2A1 in T lymphocytes. PLoS One 11: e0159714. [Crossref]
- Luz-Crawford P, Noël D, Fernandez X, Khoury M, Figueroa F, et al. (2012) Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PLoS One 7: e45272. [Crossref]
- Yang L, Qiao G, Hassan Y, Li Z, et al. (2016) Program death-1 suppresses autoimmune arthritis by inhibiting Th17 response. Arch Immunol Ther Exp (Warsz) 64: 417-423. [Crossref]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, et al. (2009) C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 10: 514-523. [Crossref]
- Kochi Y, Okada Y, Suzuki A, Ikari K, Terao C, et al. (2010) A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet 42: 515-519. [Crossref]
- Zhou XJ, Mu R, Li C, Nath SK, Zhang YM, et al. (2015) Association of variants in CCR6 with susceptibility to lupus nephritis in Chinese. Arthritis Rheumatol 67: 3091–3093. [Crossref]
- Koga T, Otomo K, Mizui M, Yoshida N, Umeda M, et al. (2016) Calcium/Calmodulin-dependent kinase IV facilitates the recruitment of interleukin-17-producing cells to target organs through the CCR6/CCL20 Axis in Th17 cell-driven inflammatory diseases. Arthritis Rheumatol 68:1981-1988. [Crossref]
- Elhofy A, Depaolo RW, Lira SA, Lukacs NW, Karpus WJ (2009) Mice deficient for CCR6 fail to control chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 213: 91-99. [Crossref]
- Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, et al. (2012) Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61:1693-1700. [Crossref]
- Patel DD, Lee DM, Kolbinger F, Antoni C (2013) Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis 72 Suppl 2: ii116-123. [Crossref]
- Yan L, Yang C, Tang J (2013) Disruption of the intestinal mucosal barrier in Candida albicans infections. Microbiol Res 168: 389-395. [Crossref]