Abstract
Purpose: To understand the cellular and molecular basis of the association between cigarette smoking and age-related macular degeneration (AMD), we examined the effect of Benzo(e)Pyrene (B(e)P), a toxic element in cigarette smoke, on a Müller cell line (MIO-M1).
Results: . There was a significant decrease in cell viability of the MIO-M1 cells which were treated with three concentrations of B(e)P (200 μM, 100 μM and 50 μM). The increase in ROS/RNS production in comparison with their respected controls was significant for the concentrations of 200 µM, 100 µM, and 50 µM. The ΔΨМ was significantly decreased after treatment with B(e)P at 200 µM , 100 µM and 50 µM. Caspase-3/7 activity was significantly increased for cells treated with 200 μM, 100 μM, 50 μM compared to DMSO controls. There was marked DNA fragmentation in cells exposed to B(e)P 200 µM.
Conclusions: B(e)P causes oxidative damage and apoptosis of MIO-M1 cells. Müller cells function as primary glial cells. They also have the potential to undergo dedifferentiation into multipotent progenitor cells. Disruption of these important functions by B(e)P may contribute to the significant risk that smoking has for development of AMD.
Key words
Benzo(e)Pyrene, müller cells, smoking, AMD, apoptosis
Abbreviations
AMD: Age Related Macular Degeneration; B(e)P: Benzo(e)Pyrene; RPE: Retinal Pigment Epithelial; HMVEC: human microvascular endothelial cells; MIO-M1: Human Muller Cells; ROS/RNS: Reactive Oxidation Species/Reactive Nitrogen Species; DMSO: Dimethyl Sulfoxide; H2DCFDA: 2’,7’-dichlorodihydrofluorescein diacetate; ΔΨm: Mitochondrial Membrane Potential.
Introduction
Age-related macular degeneration (AMD) is a very common cause of blindness in the aged population of the Western world [1]. One of the risk factors which is statistically associated with both the dry and the wet forms of macular degeneration is smoking [2]. In the wet form of AMD there is an abnormality in the vascular cell proliferation resulting in choroidal neovascularization, neovascular membrane formation and leakage. In contrast, the dry form can have extensive retinal pigment epithelial (RPE) cell damage which can lead to geographic atrophy. Both forms of AMD have devastating effects on central vision [3]. AMD is a major cause of vision loss in the elderly American population and the prevalence of AMD is currently estimated to be 1.75 million in United States [4].
It is well known that in AMD, there is damage to multiple cell types within the macula. One of the primary sites of damage in AMD is at the level of RPE cells and Bruch's membrane, but in addition, death of photoreceptors and inner nuclear layer cells have been demonstrated in studies on postmortem eyes in AMD patients [5]. Müller cells are the primary glial cells of the human retina, nourishing retinal neurons. They also deactivate and recycle neurotransmitters and maintain the ionic homeostasis within the retinal environment. Another interesting feature of these cells is their potential to undergo dedifferentiation into multipotent progenitor cells. These can in turn subsequently differentiate into other retinal cell types such as photoreceptors and RPE cells if they are damaged during a disease process [6]. Therefore Müller cells have an extremely important role in maintaining a healthy retina and it is likely they are involved either directly or indirectly in AMD pathology. Exploring the effects of B(e)P upon the human Müller cell line, MIO-M1, is important for clarification of the role that cigarette smoke toxins might play in retinal degenerations.
The etiology of AMD is not well defined but it is believed to be multifactorial. Both genetic and environmental factors are considered to play a role in its pathogenesis [7]. Cigarette smokers have been found to have a two-fold to three-fold increase in the incidence of AMD compared to non smokers [8]. Cigarette smoke contains injurious polycyclic aromatic hydrocarbons (PAH) [9] such as Benzo(a)pyrene (B(a)P) and Benzo(e)pyrene (B(e)P) which have been shown to be toxic to RPE cells in vitro [3,10]. Multitudes of other toxins have been shown to be toxic to retinal cells in the in vitro studies. Patil et al. showed that nicotine was toxic to human microvascular endothelial cells (HMVEC) and rat neurosensory retinal (R28) cells. They proposed that the mechanism of toxicity for the R28 cells was through oxidative pathway which was non-caspase, non-calpain mediated. The toxicity in case of HMVEC cells was via necrosis [11].
Caspases are part of a family of cysteine proteases which are required in cells for apoptosis which is programmed cell death. This apoptotic process can occur in response to injury or developmental events and leads to cellular disassembly and death. In humans, eleven caspases have been identified. Caspases are normally present in an inactive stage and can be divided into two types-the initiator (apical) and effector (executioner) forms. The initiator caspases (caspase-2, -8, -9, -10 and -12) activate the effector caspases (Caspase-3, -6, -7) by cleaving their inactive proforms into active forms [12]. Caspase-3/7 activation is a hallmark of apoptosis and represents the final commitment to cellular disassembly.
In the present study we evaluated the effects of B(e)P on the human MIO-M1 cell line, which is representative of Müller glial cells of the human retina. In addition to studying the toxicity, we also evaluated the pathways and mechanisms through which B(e)P acts. We found that MIO-M1 cells treated with different concentrations of B(e)P showed decreased cell viability, increase in caspase 3/7 activity, increase in ROS/RNS production and a decrease in mitochondrial membrane potential.
Materials and methods
Cell culture
The human MIO-M1 (Moorfields/Institute of Ophthalmology-Müller 1) cell line was obtained from Dr. Astrid Limb of the Department of Cell Biology and Pathology, Institute of Ophthalmology and Moorsfield Eye Hospital, London, United Kingdom. MIO-M1 is a spontaneously immortalized Müller cell line [13]. Cells that were used for these series of experiments were of passage 26 to 30. The cells were grown in 75 cm2 tissue culture flasks until confluent. The culture medium was D-MEM containing 4500 mg/L glucose (Invitrogen Gibco, Carlsbad, Ca), L-glutamine, 10% fetal bovine serum (heat inactivated, Invitrogen), and penicillin/streptomycin (Invitrogen). When the cells were confluent, they were cultured in 2% serum culture medium to convert them to a non proliferative stage. This was done to simulate the condition in which the Müller cells exist in the human eyes. For the cell viability studies, 5.5 × 105 cells were placed in 6-well plates (Becton Dickinson Labware, Franklin Lakes, NJ). For JC-1, ROS and Caspase 3/7 assays, 24-well plates were used and 1.5 × 105 cells per well were plated. Experiments were carried out in triplets and the entire experiment was repeated two times. All the nine values were then fed into the statistical software for analysis.
Exposure to B(e)P
B(e)P was obtained commercially in powder form. From the 0.0252 gram of B(e)P powder received, a stock solution of 100 mM was prepared by dissolving 0.0252 gram of B(e)P in 1 ml DMSO (Dimethyl Sulfoxide). From this, four concentrations of 200 µM, 100 µM, 50 µM and 25 µM were prepared. The cells were treated for 24 hours with the four concentrations of B(e)P along with the equivalent concentrations of DMSO (which served as controls).
Cell viability assay
The cell viability assay was performed with the use of an automated analyzer (Vi cell; Beckman Coulter, Fullerton, CA) using the technique described by Narayanan et al. [14] The cells were washed with PBS and then detached from the 6-well plates using 0.2% trypsin. Cells were centrifuged at 1000 rpm for 5 min and then resuspended in 1 ml culture medium. The automated analyzer was then used which works on the principle of automated trypan blue dye-exclusion assay. The cell viability was obtained in terms of percentage of viable cells out of total number of cells.
Detection of reactive oxygen species/reactive nitrogen (ROS/RNS) species
ROS/RNS accumulation in tissues is damaging to mitochondrial DNA and causes mitochondrial dysfunction [15]. Fluorescent dye 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA; Invitrogen) was used to measure the ROS/RNS production. It measures hydrogen peroxide, hydroxyl radicals and peroxynitrite anions. The MIO-M1 cells were plated in 24-well plate and then incubated 24 hours until confluent. The cells were then treated with the four concentrations of B(e)P along with corresponding DMSO controls. B(e)P treated cells were incubated for 24 hours, washed with PBS and incubated with 10 μM H2DCFDA for 30 min. The ROS/RNS levels were measured using the scanning unit FMBIO III from Hitachi with excitation wavelength of 488 nm and emission wavelength of 520 nm.
Mitochondrial membrane potential (ΔΨm) measurements
The mitochondrial membrane potential was measured using a JC-1 assay kit (Cell Technology, Minneapolis, MN). The assay is based upon a cationic dye (5,5’6,6’-tetrachloro-1 to 1’,3,3’-tertaethylbenzimidazolylcarbocyanine iodide). In healthy cell, the dye enters the mitochondria and fluoresces red. In apoptotic cells, the ΔΨm collapses so that the dye remains in the cytoplasm and fluoresces green. The ratio of red to green fluorescence is calculated with the ratio being higher in healthy cells. 100,000 MIO-M1 cells were plated in each well in 0.5 ml culture medium, cultured at 37°C in 5% CO2 for 24 hours until confluent. The cultures were treated with 200 µM, 100 µM, 50 µM or 25 µM of B(e)P. The controls were DMSO-equivalent cultures or untreated cultures. The B(e)P treated cells were cultured at 37°C for 24 hours and then exposed to the JC-1 reagent for 15 min. Cells were washed with PBS and fluorescence was quantified using a fluorescence image scanning instrument at 532-nm excitation and 580-nm emission wavelengths (red fluorescence) and 488-nm excitation and 520-nm (green fluorescence). The red to green ratios were calculated.
Caspase-3/7 detection
Levels of caspase-3/7 activity are an indicator of apoptosis. The carboxyfluorescein FLICA Apoptosis Detection Kit (Immunochemistry Technologies LLC, Bloomington, MN) was used wherein the FLICA probes bind to caspase-3/7 and emit fluorescence in the range of 515 to 535 nm and are excited in the range of 488 to 492 nm. The cells which do not undergo apoptosis have no FLICA probes attached to them and hence they do not show fluorescence. The cells undergoing apoptosis fluoresce brightly. The level of apoptosis as measured by caspase-3/7 activity is quantified by the degree of fluorescence emitted by the FLICA probes.
The methodology of the experiment was as follows. After B(e)P treatment, cells were washed with culture medium which was then replaced with 1 X FLICA solution in 300 µl of culture medium and incubated for 1 hour at 37°C. The controls consisted of cells treated with DMSO-equivalent cultures and untreated cell cultures, wells without cells with buffer alone and wells without cells but with culture medium and DMSO. The quantification of fluorescence was performed using a fluorescence image scanning instrument (FMBIO III from Hitachi). The average signal intensity of the fluorescence of the pixels in a spot represents the caspase-3/7 activity.
DNA fragmentation assay
Cells were plated in 6-well plate and incubated for 24 hours till they were confluent. Then they were exposed to B(e)P for 24 hours in serum free media. DNA of the cells was extracted using the QIAamp DNA Micro kit (Qiagen, Hilden, Germany). The DNA samples were subjected to electrophoresis on 3% agarose gel which had been pre-treated with 5% ethidium bromide to stain the DNA samples. A 100 bp marker was used in the first ladder. Imaging of the stained gel was performed using the scanning unit FMBIO III from Hitachi.
Statistical analysis
The data gathered were analyzed by ANOVA (Prism, version 3.0; GraphPad Software, San Diego, California). The data within each experiment was compared using the Newman-Keuls multiple-comparison test. P values less than 0.05 were considered statistically significant. The error bars in the graphs signify standard error mean of experiments done in triplicate.
Results
Cell viability assay
Mean cell viability of MIO-M1 cells treated with B(e)P with concentrations 200 μM, 100 μM, 50 μM and 25 μM was 75.13 ± 1.520, 84.30 ± 0.1581, 87.18 ± 0.6933 and 89.25 ± 1.479, respectively (Figure 1). The mean cell viabilities of DMSO equivalent controls were 86.98 ± 0.4871, 90.28 ± 0.5498, 90.80 ± 0.2799 and 91.28 ± 0.3038, respectively, and they were very similar to the mean cell viability of untreated control which was 94.23 ± 0.4289. There was a significant dose-dependent decrease in cell viability of the MIO-M1 cells which were treated with higher three concentrations of B(e)P (200 μM, 100 μM and 50 μM).
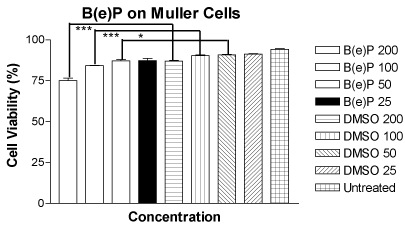
Figure 1. Figure 1. Bar graph showing a significant decrease in cell viability in MIO-M1 cells after 24 hours treatment with B(e)P 200 µM, 100 µM, and 50 µM compared to respective DMSO-equivalent controls. B(e)P 25 µM-treated cultures showed similar cell viability compared to DMSO-equivalent control and untreated cells. B(e)P, Benzo(e)Pyrene; DMSO, Dimethylsulfoxide. ***p<0.001. *p<0.05.
ROS/RNS assay
The mean relative fluorescence values for the MIO-M1 cells treated with four concentrations of B(e)P of 200 μM, 100 μM, 50 μM and 25 μM were 2835 ± 190.0, 2704 ± 111.7, 2339 ± 76.03 and 2274 ± 67.57, respectively (Figure 2). The mean relative fluorescence values for the MIO-M1 cells treated with equivalent four DMSO equivalent controls were 1199 ± 36.09, 1193 ± 49.26, 1369 ± 28.98 and 1281 ± 26.36, respectively. The relative fluorescence of the untreated cells was 678.8 ± 30.65. The increase in ROS/RNS production in comparison with their respected controls was significant for the concentrations of 200 µM, 100 µM, and 50 µM.
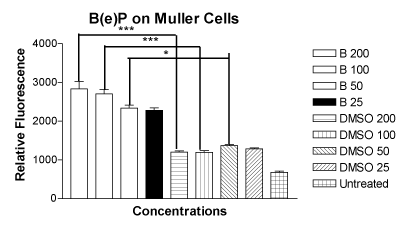
Figure 2. Bar graph showing significantly increased ROS/RNS levels in MIO-M1 cells after treatment for 24 hours with B(e)P 200 µM, 100 µM, and 50 µM compared to respective DMSO-equivalent controls. Untreated, B(e)P 25 µM and equivalent DMSO cultures showed low ROS/RNS levels. B(e)P, Benzo(e)Pyrene; DMSO, Dimethyl sulfoxide. ***p<0.001. *p<0.05.
Mitochondrial membrane potential (ΨΔm) assay
The mean values of the ratio of red to green fluorescence in the JC-1 assay for the MIO-M1 cells treated with four concentrations of B(e)P of 200 μM, 100 μM, 50 μM and 25 μM were 3.248 ± 0.1383, 3.962 ± 0.1442, 5.158 ± 0.1855 and 5.615 ± 0.1500, respectively (Figure 3). The mean values of the ratio of red to green fluorescence for the cells treated with four DMSO equivalent controls were 6.642 ± 0.1474, 6.440 ± 0.2243, 6.783 ± 0.1418 and 6.818 ± 0.1514, respectively. The mean value for the untreated cells was 8.383 ± 0.2330. There was a significant alteration of ΨΔm values in MIO-M1 cells treated with B(e)P concentrations of 200 µM, 100 µM, and 50 µM.
2021 Copyright OAT. All rights reserv
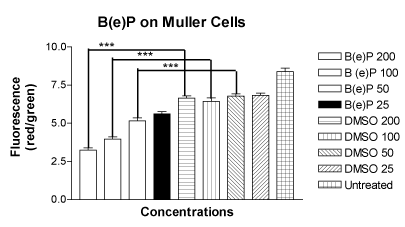
Figure 3. Bar graph showing significantly low red/green fluorescence in MIO-M1 cells after treatment for 24 hours with B(e)P 200 µM, 100 µM, and 50 µM compared to respective DMSO-equivalent controls. Untreated, B(e)P 25µM and equivalent DMSO cultures did not show significantly low red/green ratio. B(e)P, Benzo(e)Pyrene; DMSO, Dimethyl sulfoxide. ***p<0.001.
Caspase-3/7 activity
The mean values of the caspase-3/7 activity for the MIO-M1 cells treated with B(e)P at 200μM, 100μM, 50μM and 25 μM were 9804 ± 150.4, 8999 ± 146.5, 6864 ± 283.7 and 4592 ± 193.4, respectively (Figure 4). The mean values of the caspase-3/7 activity for the cells treated with equivalent four DMSO controls were 4113 ± 80.50, 2619 ± 232.3, 2559 ± 211.9 and 2535 ± 254.2, respectively. The mean caspase-3/7 activity value for the untreated cells was 347.3 ± 87.96. The rise in caspase-3/7 activity was significant for the MIO-M1 cells treated with 200 µM, 100 µM, and 50µM concentrations of B(e)P.
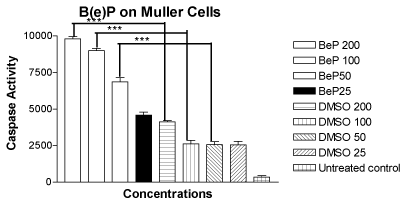
Figure 4. Bar graph showing significantly increased caspase-3/7 activity in MIO-M1 cells after 24 hours treatment with B(e)P 200 µM, 100 µM, and 50 µM compared to respective DMSO-equivalent cultures. Untreated, B(e)P 25 µM and equivalent DMSO cultures showed minimal caspase-3/7 activity. B(e)P, Benzo(e)Pyrene; DMSO, Dimethylsulfoxide. ***p<0.001.
DNA laddering for apoptosis
The DNA extracted from the cells exposed to B(e)P 200 µM showed marked DNA fragmentation as compared to the control (Figure 5). The DNA fragmentation appeared at 200 bp intervals.
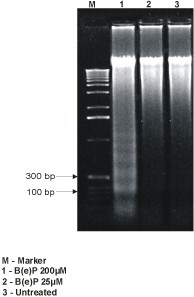
Figure 5. DNA fragmentation analysis showing bands at 200-bp interval in 50 µM-B(e)P treated MIO-M1 cultures.
Discussion
Cigarette smoking is a well-known risk factor for AMD but the mechanism of it in AMD progression is gradually being elucidated. The lab has been working on this vexing problem for the past five years. Some crucial pathways have been uncovered that explain the cell death which could probably be playing a major role in aging human eyes. Cigarette smoke is a mixture of various chemicals and they are well known to have a toxic effect on several human cells. The numbers of chemical compounds in a cigarette smoke are known to be over 4000 [16]. Toxins of cigarette smoke are known to cause oxidation of important molecules which play a role in cellular growth and turn over [17].
In AMD, multiple cell types within the macula can be damaged. While the primary pathology involves the RPE cells, Bruch’s membrane, choriocapillaries [18] and other cell types of the overlying neuroretina are also affected. RPE cells have been extensively studied by previous researchers from our lab using different cigarette toxins [3,11]. In this study the focus was on the retinal glial cells. Müller cells are the predominant glial cells in retina. They play crucial role in functioning and well-being of photoreceptors and other retinal neurons. Previous studies have shown toxicity of B(e)P on retinal pigment epithelial cells (RPE) but no work has been done so far on Müller cells. In this study MIO-M1 cells were taken to be a representative model of the glial cells of the entire retina and were evaluated for the effect of a component of cigarette smoke on it. This study showed a significant decrease in cell viability of the MIO-M1 cells which were treated with B(e)P at the three concentrations of 200 μM, 100 μM and 50 μM. This shows significant percentage of cell death as compared to their DMSO controls which rules out the possibility of the death due to DMSO.
Human retina has three basic types of glial cells: Muller cells, astroglia and microglia. Müller cells provide a supportive function to the neurons of retina including the photoreceptors, bipolar cells and ganglion cells [19]. They also preserve the homeostasis of the retina by secreting growth factors and cytokines that maintain the outer blood-retinal barrier [20]. In AMD, there is a loss of RPE cell barrier function. This barrier dysfunction is believed to lead to accumulation of lipid bloated micro-glial cells in the subretinal spaces [21]. These lipid bloated cells on fundoscopy appear to look very similar to the drusen. With loss of the RPE cell barrier, choroidal neovascularization can occur. The Müller cells may be involved indirectly in the pathology of AMD since they are supportive of the outer blood-retinal barrier which is damaged both in the early and late stages of AMD.
Recent studies have reported the progenitor properties of Müller cells. Müller cells in isolation display features very similar to retinal progenitor cells. They can renew themselves and generate all of the neuronal cell types characteristic of retina [22,23]. Following retinal degeneration, progenitor cells derived from Müller cells differentiate into several different retinal cell lineages which can support retinal regeneration in vivo [24,25]. However, because they are limited in numbers, these new retinal cells cannot completely replace damaged tissues [25]. Even after retinal degeneration has occurred, Müller cells continue to function to minimize collateral damage [26,27]. This includes their re-entering the cell cycle and producing neuroprotective growth factors. Once these cells are activated, they become involved in the formation of glial scar which occurs in late phases of retinal degeneration [26]. The proliferation of Müller cells is regulated by the Wnt/β-catenin (Wnt) pathway and the sonic hedgehog (Shh) pathway [28].
In living organisms, the cells are kept at a reduced environment. This is done through the means of enzymes. Oxidants are constantly produced during the body metabolism, one example being the generation and leakage of activated oxygen from mitochondria during the oxidative phosphorylation. Oxidative stress can be defined as the imbalance between the production of free or reactive oxygen and the ability of the organism or cells to detoxify the oxidants and their intermediatories or the damage that ensues from it [29]. Oxidants from the cigarette smoke toxins could be increasing this insult. This can be occurring by at least two ways. One by the presence of oxidative chemicals in the cigarette smoke itself. The second contributing factor could be damage to the mitochondria by the toxins hence decreased phagocytosis of the free radicals by the mitochondrial machinery. Chemically, oxidative stress translates as sharp rise in the cellular reduction potential, that is, it becoming less negative. One of the aspects of oxidative stress is the generation of reactive oxidative species such as peroxides and free radicals. Oxidative stress is known to trigger apoptosis and in more severe cases, even necrosis [30]. In response to oxidative stress, cells release various inflammatory mediators and cytokines like interleukins (ILs) and tumor necrosis factor (TNF) [31]. Production of the reactive oxygen species (ROS) is a particular destructive effect of oxidative stress. The generation of ROS was tested by the assay and a significant increase in ROS/RNS production was found for the concentrations of 200 µM, 100 µM, and 50 µM in comparison with their respected controls.
Apoptosis is a well known form of cell death that occurs in organisms both physiologically and pathologically. In physiologic form, apoptosis is a tool for cell replacement of damaged cells and tissue remodeling. Apoptotic cells exhibit shrinkage of cells, chromatin condensation and DNA fragmentation. It is known to occur in response to oxidative stress [32].
One of the objectives of this study was to look at the early events in apoptosis. Decrease in mitochondrial membrane potential is a hallmark of early apoptosis. JC-1 kit was used to measure the ΨΔm. A significant alteration of mitochondrial membrane potential in MIO-M1 cells were found that were treated with B(e)P concentrations of 200 µM, 100 µM, and 50 µM as compared to their respective controls signifying early apoptosis.
To know if late apoptosis sets in after 24-hour of drug exposure, the levels of caspases 3/7 were evaluated. A significant rise in caspase activity was found for the MIO-M1 cells treated with 200 µM, 100 µM, and 50 µM concentrations of B(e)P. This suggested that within 24-hour duration, late apoptosis had set in and this probably was ultimately responsible for the decrease in cell viability that was found. DNA laddering is another technique used to denote apoptosis as it shows the genotoxic damage occurring in apoptotic cells. The DNA fragmentation confirms the late apoptosis occurring within the chromatin of the MIO-M1 cells after exposure to B(e)P.
In this in vitro study, it has shown quite comprehensively that the cigarette smoke compound, BeP, is able to induce a combination of two insults to the cells, both oxidative and apoptotic, either sequentially or temporally independently. Both oxidation and apoptosis were dose-dependent. It is possible that that the oxidative and apoptosis pathways either run in parallel or in series whereby the oxidative stress induces apoptosis. Based on this, it can be speculated that the combination of oxidative stress and apoptosis which was observed in cell lines in these vitro studies may be occurring many thousands of times in a smoker's life either as repetitive solo events or in cumulative fashion. This could in turn lead to the death of Müller cells and thereby to an impairment of their functioning in the repair and support of RPE cells and photoreceptors. This could be a compounding factor for the damage to the RPE cells and photoreceptors in AMD patients who smoke. Age would be the primary factor for their decreased functioning.
The role of antioxidants becomes important when one considers the outcome of these experiments. Questions will remain as to how much to give, when to start and the role of diet in the pathogenesis of AMD. Intraocular modes of delivery of anti oxidants can be looked upon as a better way of drug delivery.
Supported by
The Discovery Eye Foundation, the Henry L. Guenther Foundation, the Iris and B. Gerald Cantor Foundation, Ko Family Foundation, the Lincy Foundation, and Unrestricted grant from Research to Prevent Blindness Foundation.
References
- Fine SL, Berger JW, Maguire MG, Ho AC (2000) Age-related macular degeneration. N Engl J Med 342: 483-492. [Crossref]
- Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, et al. (2005) Smoking and age-related macular degeneration: a review of association. Eye (Lond) 19: 935-944. [Crossref]
- Sharma A, Neekhra A, Gramajo AL, Patil J, Chwa M, et al. (2008) Effects of Benzo(e)Pyrene, a toxic component of cigarette smoke, on human retinal pigment epithelial cells in vitro. Invest Ophthalmol Vis Sci 49: 5111-5117. [Crossref]
- Friedman DS, O'Colmain BJ, Muñoz B, Tomany SC, McCarty C, et al. (2004) Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 122: 564-572. [Crossref]
- Dunaief JL, Dentchev T, Ying GS, Milam AH (2002) The role of apoptosis in age-related macular degeneration. Arch Ophthalmol 120: 1435-1442. [Crossref]
- Bernardos RL, Barthel LK, Meyers JR, Raymond PA (2007) Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J Neurosci 27: 7028-7040. [Crossref]
- Tezel TH, Bora NS, Kaplan HJ (2004) Pathogenesis of age-related macular degeneration. Trends Mol Med 10: 417-420. [Crossref]
- Tomany SC1, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, et al. (2004) Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology 111: 1280-1287. [Crossref]
- Vakharia DD, Liu N, Pause R, Fasco M, Bessette E, et al. (2001) Polycyclic aromatic hydrocarbon/metal mixtures: effect on PAH induction of CYP1A1 in human HEPG2 cells. Drug Metab Dispos 29: 999-1006. [Crossref]
- Patton WP, Routledge MN, Jones GD, Lewis SE, Archer DB, et al. (2002) Retinal pigment epithelial cell DNA is damaged by exposure to benzo[a]pyrene, a constituent of cigarette smoke. Exp Eye Res 74: 513-522. [Crossref]
- Patil AJ, Gramajo AL, Sharma A, Seigel GM, Kuppermann BD, et al. (2009) Differential effects of nicotine on retinal and vascular cells in vitro. Toxicology 259: 69-76. [Crossref]
- Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 68: 383-424. [Crossref]
- Limb GA, Salt TE, Munro PM, Moss SE, Khaw PT (2002) In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Invest Ophthalmol Vis Sci 43: 864-869. [Crossref]
- Narayanan R, Kenney MC, Kamjoo S, Trinh TH, Seigel GM, et al. (2005) Trypan blue: effect on retinal pigment epithelial and neurosensory retinal cells. Invest Ophthalmol Vis Sci 46: 304-309. [Crossref]
- Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 94: 514-519. [Crossref]
- Church DF, Pryor WA (1985) Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 64: 111-126. [Crossref]
- Repine JE, Bast A, Lankhorst I (1997) Oxidative stress in chronic obstructive pulmonary disease. Oxidative Stress Study Group. Am J Respir Crit Care Med 156: 341-357. [Crossref]
- Zarbin MA (2004) Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol 122: 598-614. [Crossref]
- Newman E, Reichenbach A (1996) The Müller cell: a functional element of the retina. Trends Neurosci 19: 307-312. [Crossref]
- Constable PA, Lawrenson JG (2009) Glial cell factors and the outer blood retinal barrier. Ophthalmic Physiol Opt 29: 557-564. [Crossref]
- Combadière C, Feumi C, Raoul W, Keller N, Rodéro M, et al. (2007) CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest 117: 2920-2928. [Crossref]
- Das AV, Mallya KB, Zhao X, Ahmad F, Bhattacharya S, et al. (2006) Neural stem cell properties of Müller glia in the mammalian retina: regulation by Notch and Wnt signaling. Dev Biol 299: 283-302. [Crossref]
- Kubota A, Nishida K, Nakashima K, Tano Y (2006) Conversion of mammalian Müller glial cells into a neuronal lineage by in vitro aggregate-culture. Biochem Biophys Res Commun 351: 514-520. [Crossref]
- Ooto S (2006) [Potential for neural regeneration in the adult mammalian retina]. Nippon Ganka Gakkai Zasshi 110: 864-871. [Crossref]
- Osakada F, Ooto S, Akagi T, Mandai M, Akaike A, et al. (2007) Wnt signaling promotes regeneration in the retina of adult mammals. J Neurosci 27: 4210-4219. [Crossref]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, et al. (2006) Müller cells in the healthy and diseased retina. Prog Retin Eye Res 25: 397-424. [Crossref]
- Burke JM, Smith JM (1981) Retinal proliferation in response to vitreous hemoglobin or iron. Invest Ophthalmol Vis Sci 20: 582-592. [Crossref]
- Wan J, Zheng H, Xiao HL, She ZJ, Zhou GM (2007) Sonic hedgehog promotes stem-cell potential of Müller glia in the mammalian retina. Biochem Biophys Res Commun 363: 347-354. [Crossref]
- MacNee W (2000) Oxidants/antioxidants and COPD. Chest 117: 303S-17S. [Crossref]
- Lennon SV, Martin SJ, Cotter TG (1991) Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif 24: 203-214. [Crossref]
- Rahman I, MacNee W (1998) Role of transcription factors in inflammatory lung diseases. Thorax 53: 601-612. [Crossref]
- Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239-257. [Crossref]