Abstract
A dysmorphic neonate is a cause of concern and anxiety for the parents and the physician. It allows a more accurate estimate of the risk of recurrence and therefore allows genetic counselling. It allows prognostication and permits interventions that may prevent, anticipate or more successfully treat complications.TheAround 4,000 malformation syndromes have now been delineated and many are associated with medical problems. Thus making a specific syndrome diagnosis can influence immediate medical management. A detailed history, a physical examination for detailing the major and minor anomalies, recording the growth, examination of previous records and photographs are complemented by cytogenetics and molecular genetic techniques in achieving a diagnosis. Familiarity with dysmorphology databases and cross referencing the anomalies especially the rarer ones helps in narrowing the differential diagnosis.
Key words
Dysmorphism, Malformation, Deformation, Sequence, Association, Syndrome, Disruption, Dysplasia, Anomaly, Karyotyping, FISH, DNA microarray, molecular analysis
Introduction
Dysmorphology is a branch of clinical genetics in which clinicians and researchers study and attempt to interpret the patterns of human growth and structural defects. A dysmorphic neonate at birth is a cause of concern and anxiety for the parents and also the physician. If the dysmorphism does not result in a major anomaly and the parents are unaware of the presence of a dysmorphic syndrome in their newborn, breaking the news, bringing the variations to the notice of the parents, characterizing all major and minor anomalies accurately, establishing and confirming the diagnosis and providing genetic counselling are the immediate responsibilities of the physician. Providing health care supervision for the child on follow-up as per the established guidelines is subsequently required and can be done accurately only if the diagnosis is confirmed. With the increasing identification of genetics in the causation of disease, reaching a clinical diagnosis also allows a more targeted search for a genetic aetiology. Making a diagnosis also allows the parents to search for and join a ‘support group’ and interact with other parents with children having the same or similar problems. This helps the parents to cope with stress of having a dysmorphicbaby. The approach to making a diagnosis of a dysmorphic neonate is fundamentally similar to making a diagnosis of a neonate with any systemic illness and relies on a detailed history, a meticulous clinical examination, identifying a syndrome based on a combination of signs, or sometimes ‘by gestalt’ (pattern recognition). Cytogenetics and molecular techniques improve our ability to make precise syndrome diagnoses. Eventhough there is a certain degree of urgency in making a diagnosis in a dysmorphic neonate, a snap diagnosis should never be made, as experience teaches us that in most cases they are wrong and an infant could carry on with a wrong diagnosis for many months before it may get corrected [1].
The Basic Concepts of Dysmorphology
The word ‘dysmorphic’ originates from a greek word ‘dys’ meaning disordered and ‘morph’ meaning shape or form. The term ‘dysmorphology’ was coined by Dr David Smith in 1960’s in USA. Around one in 40 or 2.5% of all newborns have a malformation at birth. This may be an isolated malformation or may occur together with other malformations and/or dysmorphic features as part of a malformation syndrome. Around 4,000 malformation syndromes have now been delineated. Many are associated with medical problems and making a specific syndrome diagnosis can influence immediate medical management. However, the child with dysmorphism often does not have a major malformation, and may simply have an appearance that is unusual compared with the general population and of unaffected close relatives [2].Most syndromes occur due to one of the following causes:-
- Single gene disorders e.g. Apert syndrome
- Chromosomal disorders e.g. Down syndrome
- Microdeletion syndromes e.g. Prader-Willi syndrome
- Polygenic disorders e.g. club foot
- Environmental causes (Teratogenesis) e.g. Rubella, congenital viral infection, infant of diabetic mother (IDM)
Some inborn errors of metabolism may result in dysmorphism e.g. peroxisomal disorders (Zellweger syndrome), pyruvate dehydrogenase deficiency, glutaricaciduria type II. These are progressive in nature with worsening of the dysmorphism with time [3].Syndromes due to chromosomal anomalies are non-progressive but the phenotype may become more explicit with time e.g. Smith-Magenis syndrome.1A genetic etiology should be suspected if the infant has any of the following :-[4]
- Congenital anomalies: one or more major anomaly or more than two minor anomalies
- Poor growth: symmetric intrauterine growth restriction or postnatal growth failure
- Developmental delay or developmental regression
- Craniofacial dysmorphism
- Ambiguous genitalia
A basic knowledge of the embryologic and fetal development is integral to understanding of the susceptibility to dysmorphism which is highlighted in the Figure 1 [5].Critical periods during the development of the fetal organs during which teratogenic influences lead to anomalies and dysmorphism can be understood from Figure 2.[6]The definition of terms commonly used in description of birth defects is as given below in Table 1. [2, 7]Syndrome diagnosis depends upon good clinical skills, knowledge of the description and definition of human phenotypic variations, experience of the examiner supported by dysmorphology databases and cytogenetics and molecular genetic studies. There is undoubtedly an element of ‘intuition’ which is unteachable and this ability combined with reading and recall results in the ‘edge’ that some clinicians have over others.1 The definition of common clinical signs seen in dysmorphic syndromes were neatly outlined ironing out ambiguous or unclear references through the efforts of a group of over 30 clinical geneticists from the United States, Canada, Europe, and Australia. These consensus definitions have been published in the ‘American journal of clinical genetics’ in 2008. [8-14]The reader is encouraged to look at this reference to improve his or her understanding of these signs. A similar but shorter and less exhaustive list is given in Table 2.[7]It is also useful to categorize abnormalities as ‘major’ or ‘minor’ birth defects. Major anomalies are those that either cause dysfunction (absence of a digit) or require surgical correction (polydactyly), while minor anomalies which occur in less than 5% of the population neither cause significant dysfunction nor require surgical correction (mild cutaneous syndactyly). The significance of identifying the ‘minor’ anomalies is that they herald the increased risk or the presence of a ‘major’ anomaly as shown below in Table 3. [15]A list of minor anomalies seen in various systems is as given below in Table 4.[7]The diagnosis of the dysmorphic infant therefore requires a methodical approach beginning with history with emphasis on the perinatal, family and exposure history and re-examination of the neonate and ordering investigations based on the this evaluation. It requires a basic knowledge of the common clinical signs in dysmorphic syndromes (human phenotypic variations) and knowledge of the major and minor anomalies. It requires an element of clinical suspicion which improves with reading and following case reports and case series published in achieved journals. It requires an instinct and a particular aptitude. There is no substitute for the repeated observation of important clinical signs and as the clinical signs suggesting specific diagnoses are rarely encountered, repeated review of literature is necessary.
|
Terminology
|
Meaning
|
|
Malformation
|
morphologic abnormality that arises because of an abnormal developmental process. A primary error in morphogenesis e.g. cleft lip.
|
|
Malformation sequence
|
pattern of multiple defects resulting from a single primary malformation
e.g. talipes and hydrocephalus can result from a lumbar neural tube defect.
|
|
Malformation syndrome
|
pattern of features, often with a unifying underlying cause, that arises from several different errors in morphogenesis e.g. trisomy 21
|
|
Deformation
|
distortion by a physical force of an otherwise normal structure e.g. club foot.
|
|
Disruption
|
destruction of a tissue that was previously normal e.g. amniotic band sequence
|
|
Dysplasia
|
abnormal cellular organisation within a tissue resulting in structural changes e.g. within cartilage and bone in skeletal dysplasias.
|
|
Association
|
group of anomalies that occur more frequently than would be expected by chance alone but that do not have a predictable pattern or unified etiology
e.g. VACTERL association.
|
Table 1: The definition of terms commonly used in description of birth defects.
|
Sign
|
Definition
|
|
Brachycephaly
|
A condition in which head shape is shortened from front to back along the sagittal plane; the skull is rounder than normal
|
|
Brachydactyly
|
A condition of having short digits
|
|
Brushfield spots
|
Speckled white rings about two thirds of the distance to the periphery of the iris of the eye
|
|
Camptodactyly
|
Permanent flexion of one or more fingers associated with missing inner phalangeal creases indicating lack of finger movement from before 8 wk of gestation
|
|
Clinodactyly
|
A medial or lateral curving of the fingers; usually refers to incurving of the 5th finger
|
|
Hypoplastic nail
|
An unusually small nail on a digit
|
|
Low-set ears
|
This designation is made when the helix meets the cranium at a level below a horizontal plane that is an extension of a line through both inner canthi
|
|
Melia
|
A suffix meaning “limb” (e.g., amelia—missing limb; brachymelia—short limb)
|
|
Ocular hypertelorism
|
Increased distance between the pupils of the two eyes
|
|
Plagiocephaly
|
A condition in which head shape is asymmetric in the sagittal or coronal plane; can result from asymmetry in suture closure or from asymmetry of brain growth
|
|
Posterior parietal hair whorl
|
A single whorl occurs to the right or left of midline and within 2 cm anterior to the posterior fontanel in 95% of cases. The whorl represents the focal point from which the posterior scalp skin was under growth tension during brain growth between the 10th and 16th wk of fetal development. Aberrant position of the whorl reflects an early defect in brain development.
|
|
Postaxial polydactyly
|
Extra finger or toe present on the lateral side of the hand or foot
|
|
Preaxialpolydactyly
|
Extra finger or toe present on the medial side of the hand or foot
|
|
Prominent lateral palatine ridges
|
Relative overgrowth of the lateral palatine ridges secondary to a deficit of tongue thrust into the hard palate
|
|
Scaphocephaly
|
A condition in which the head is elongated from front to back in the sagittal plane; most normal skulls are scaphocephalic
|
|
Shawl scrotum
|
The scrotal skin joins around the superior aspect of the penis and represents a mild deficit in full migration of the labial-scrotal folds
|
|
Short palpebral fissures
|
Decreased horizontal distance of the eye based on measurement from the inner to the outer canthus
|
|
Syndactyly
|
Incomplete separation of the fingers. It most commonly occurs between the 3rd and 4th fingers and between the 2nd and 3rd toes.
|
|
Synophrys
|
Eyebrows that meet in the midline
|
|
Telecanthus
|
Lateral displacement of the inner canthi. The inner canthal distance is increased, but the inner pupillary distance is normal.
|
|
Widow's peak
|
V-shaped midline, downward projection of the scalp hair in the frontal region. It represents an upper forehead intersection of the bilateral fields of periocular hair growth suppression. It usually occurs because the fields are widely spaced, as in ocular hypertelorism.
|
Table 2: Common clinical signs in Dysmorphic syndromes.
|
Minor anomaly
|
Significance
|
|
Occurrence of a single minor anomaly
|
15% of all newborns
3% have an associated major anomaly
|
|
Occurrence of two minor anomalies
|
Less common 0.8%
11% have an associated major anomaly
|
|
Presence of three or more minor anomalies
|
Unusual 0.5%
90% have an associated major anomaly
|
|
External minor anomalies in the head and neck region and the hand
|
Constitutes 71% of minor anomalies
|
Table 3: The significance of identifying ‘minor’ anomalies.
|
System
|
Minor anomaly
|
|
Craniofacial
|
Large fontanel
Flat or low nasal bridge
Saddle nose, upturned nose
Mild micrognathia
Cutis aplasia of scalp
|
|
Eye
|
Inner epicanthal folds
Telecanthus
Slanting of palpebral fissures
Hypertelorism
Brushfield spots
|
|
Ear
|
Lack of helical fold
Posteriorly rotated pinna
Preauricular with or without auricular skin tags
Small pinna
Auricular (preauricular) pit or sinus
Folding of helix
Darwinian tubercle
Crushed (crinkled) ear
Asymmetric ear sizes
Low-set ears
|
|
Skin
|
Dimpling over bones
Capillary hemangioma (face, posterior neck)
Mongolian spots (African Americans, Asians)
Sacral dimple
Pigmented nevi
Redundant skin
Cutis marmorata
|
|
Hand
|
Simian creases
Bridged upper palmar creases
Clinodactyly of fifth digit
Hyperextensibility of thumbs
Single flexion crease of fifth digit (hypoplasia of middle phalanx)
Partial cutaneous syndactyly
Polydactyly
Short, broad thumb
Narrow, hyperconvex nails
Hypoplastic nails
Camptodactyly
Shortened fourth digit
|
|
Foot
|
Partial syndactyly of second and third toes
Asymmetric toe length
Clinodactyly of second toe
Overlapping toes
Nail hypoplasia
Wide gap between hallux and second toe
Deep plantar crease between hallux and second toe
|
|
Others
|
Mild calcaneovalgus
Hydrocele
Shawl scrotum
Hypospadias
Hypoplasia of labia majora
|
Table 4: Minor anomalies seen in various systems.
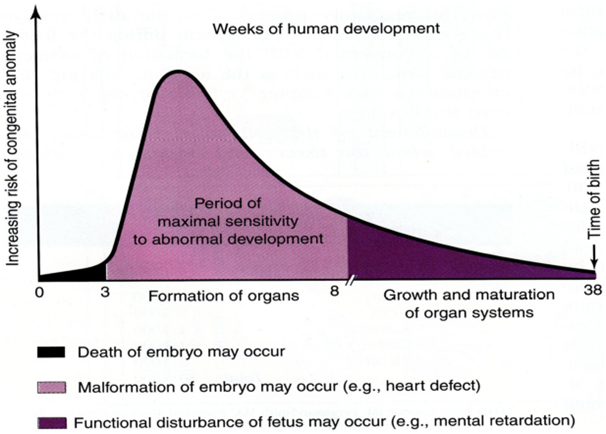
Figure 1: Embyologic and fetal susceptibility to anomalies.
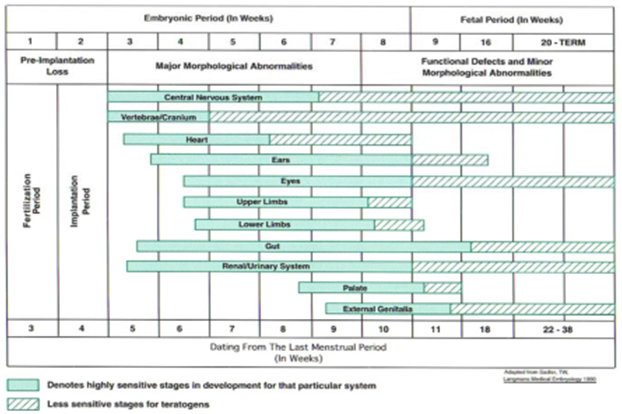
Figure 2: Critical periods during the development of the fetal organs during which teratogenic influences lead to anomalies and dysmorphisms.
The need to ‘know’
Neonates who are noted to be dysmorphic at birth carry a significantly higher risk of a major anomaly and are at a higher risk for having developmental delay. It can be an emotional roller coaster ride for a couple to know that the baby is dysmorphic, yet the diagnosis is not known and there may be a therapeutic intervention albeit rare but not being offered owing to the inability to achieve an accurate diagnosis.Making a clinical diagnosis makes it possible to attempt a targeted genetic diagnosis. It allows a more accurate estimate of the risk of recurrence and therefore allows genetic counselling. It allows prognostication and permits interventions that may prevent, anticipate or more successfully treat complications. It helps the family reach out to specific support groups and therefore helps in the coping process. [1, 16]
Approach to A Neonate With Dysmorphism
The approach to diagnosing a dysmorphic neonate is as follows:-
- History including perinatal details and family history
- Physical examination detailing the minor and major anomalies
- Growth recording and measurements
- Examination of previous records and photographs
- Making a diagnosis based on the above details :-
- iBallpark diagnosis: an ‘approximation’ of the diagnosis based upon the clinical features.
- iiDiagnosis by Gestalt: identifying the syndrome by ‘pattern recognition’.
- iiiSyndrome search: searching a dysmorphology database using the key anomalies noted in the baby
- Investigations to confirm the diagnosis and to evaluate the neonate for affected organ systems.
History
History of consanguinity suggests the possibility of an autosomal recessive disorder. Male baby with similarly affected male siblings or maternal male relatives suggests X-linked disorder. Vertical transmission suggests an autosomal dominant disorder, especially male to male transmission. History of two similarly affected siblings but of different sexes points to a possible mitochondrial disorder. History of fever with rash and polyarthralgia during pregnancy especially during the first trimester suggests a congenital infection. Drawing a three generation family tree identifies similarly affected family members and also indicates the pattern of mendelian inheritance. Recurrent miscarriages may suggest that parents are carriers of balanced chromosomal rearrangement.The occurrence of more than two first-trimester miscarriages increases the probability of finding a balanced translocation in one parent. History of maternal diabetes mellitus, acute fatty liver of pregnancy and HELLP syndrome seen in mothers carrying a fetus with a fatty acid oxidation defect should be taken.History of maternal exposure to drugs or radiation should be taken. History of alcohol or tobacco use by the mother during pregnancy should be asked. Severe hyperemesis has been linked with dysmorphic facial features and skeletal abnormalities. History of fetalakinesia or hypokinesia should be asked. The antenatal ultrasound should be examined for dating of the pregnancy, fetal growth, fetal lie and oligohydramnios or polyhydramnios. Severe oligohydramnios can predispose to congenital contractures and dysmorphic features consistent with oligohydramnios sequence (Potter syndrome). Some syndromes are associated specifically with intra-uterine growth retardation and others with fetal overgrowth. Mechanical constraint caused by uterine abnormalities e.g. bicornuate uterus can lead to fetal deformation and explain an unusual head shape or the presence of talipes deformity. Anomaly scans if available add useful information including identification of anatomic variations referred to as soft markers. But a combination of 2 or more soft markers only may indicate an underlying syndrome e.g. nuchal translucency and choroid plexus cyst. Results of maternal triple screen or amniocentesis / chorionic villus biopsy results if available should always be examined. History of intracytoplasmic fertilization (ICSI) and a large placenta indicates Beckwith-Weidemann syndrome. History of ICSI is also implicated in the causation of uniparentaldisomy 15 leading to Angelman syndrome. Therefore, history of the mode of conception – natural or assisted should be taken. Intrauterine growth restriction with discordant fetal growth from early pregnancy suggests the possibility of chromosomal anomalies, congenital malformations or fetal infection, while in-utero fetal growth restriction noted from the third trimester is mostly due to placental dysfunction and would result in an asymmetric IUGR baby. Finally an enquiry into the circumstances at the time of birth, including delayed cry, the need for resuscitation, apgar scores and whether the baby was breast feeding from day one of life should be taken. [1, 2, 7, 15, 16]
Physical Examination Detailing the Major and Minor Anomalies
A comprehensive examination detailing the major and minor anomalies must be done. A record of the weight, length, occipito-frontal circumference and ponderal index in IUGR babies at birth should be made. An examination of the dermatoglyphic patterns may occasionally be rewarding e.g. excessive arches are seen in trisomy 13, trisomy 18, Klinefelter syndrome (47XXY), cri-du-chat syndrome (5p-) and fetal phenytoin exposure. Excessive ulnar loops are seen in trisomy 21. Excessive whorls are seen in Turner syndrome (45XO), Smith-Lemli-Opitz syndrome and 18p deletion. Deltas, or triradii, form at the convergence of three sets of ridges on the palm. This junction is where the hypothenar, thenar, and distal palmar patterns converge. There are typically no triradii in the hypothenar area of the palm but when patterning is present or is large, a distal triradius arises, which is found in only 4% of normal individuals but in 85% of babies with trisomy 21.Certain clinical clues can go a long way in helping the clinician make the diagnosis e.g. hypocalcemic seizures with absent thymic shadow and a conotruncal cardiac lesion is seen in DiGeorge syndrome. Hypersensitivity to high pitched sounds, hypercalcemia, supravalvular aortic stenosis or isolated pulmonary stenosis is seen in William syndrome. A history of developmental delay, temper tantrums, occasional self-injury and sleep disturbances is seen in Smith-Magenis syndrome. [1, 2, 7, 15, 16]
Growth Recording and Measurements
Measurements such as height, weight (usually reflecting nutrition), and head circumference should be plotted on neonatal growth charts. Gestational age–appropriate charts should be used for premature infants. It is often helpful to express values that are outside the normal range as 50th percentile for a different gestational age. For example, a full-term baby with microcephaly may have a head circumference of less than the 5th percentile for 38 weeks. This can be expressed as a measurement at the 50th percentile for 33 weeks, which imparts the degree of microcephaly more clearly. Important measurements include head circumference, inner and outer canthal distances, interpupillary distances, ear length, ear placement, internipple distances, chest circumference and hand and foot lengths. Other graphs and measurements using age-appropriate standards can be found in compendia such as the Handbook of Physical measurements. [2, 17]
Examination of Previous Records and Photographs
An examination of the previous growth records would help in assessing the growth velocity and the pattern of growth. The element of subjectivity in the diagnosis of some syndromes especially at the mild end of a syndrome’s spectrum has resulted in more objective diagnostic approaches such as photogrammetry and anthropometrics. Photogrammetry uses objective measurements from standardized photographs, and anthropometry from standardized physical landmarks, to assess patients objectively.[17,18]
Making A Diagnosis
Following the history and physical evaluation, the clinician has full details about the neonate including the major and minor anomalies. If the diagnosis is not apparent based upon ‘pattern recognition’ (diagnosis by gestalt based on the examiner’s personal experience and abilities) by now, the clinician can cross reference two or more anomalies in order to create a differential diagnosis and a most-probable diagnosis (the probable diagnosis ‘approximates’ as closely as possible the true diagnosis). Narrowing the possibilities down to a few allows diagnostic testing. If there are multiple anomalies, it is prudent to use the least common ones. Cross referencing is best accomplished by using published compendia of malformation syndromes. These compendia have been supplemented by databases that are accessible online (i.e., GeneReviews, Online Mendelian Inheritance in Man (OMIM), and PubMed).[19-21]A ‘syndrome search’ can be then conducted on these online databases. The availability of such tools allows the cross referenced features to be compared easily with those of other described syndromes having similar malformations. This systematic review produces a differential diagnosis for the constellation of features described and identifies relevant literature published in this regard.
Use of Dysmorphology Databases
Databases are now considered indispensable in clinical genetics and dysmorphology. Some resources such as Online Mendelian Inheritance in Man(OMIM) have free access. It is an excellent tool for the identification of key journal references, for summaries of clinical features of classical cases and family studies of Mendelian disorders, and for obtaining an indication of the research progress in terms of gene localisation, errors therein and locus identification. Many conditions and disease genes now have dedicated databases in which all recorded mutations that cause disease are available for comparison. The identification of a variation in DNA sequence in an affected individual does not always translate into a disease-causing mutation. The existence of such databases allows clinicians to seek expert assistance from molecular genetics specialists rather than having to decide for themselves whether a DNA variation represents a ‘mutation’. It is important to be aware of the presence of such locus-specific databases. Another resource for information on rare disorders, Orphanet[22]is accessible to patients and professionals. It provides synopses and full texts on thousands of conditions, and country-specific information on specialist clinics, diagnostic laboratories and patient support groups in Europe. Another resource GeneTests[23]also contains information on testing centres as well as an encyclopaedia of information on many dysmorphic syndromes. A drawback of these resources is that there are no illustrations of the syndromes described in the databases. There are some commercial ‘expert’ systems such as London Medical Database’s, Winter–BaraitserDysmorphology Database[24] andPOSSUMweb.[25]
Investigations to Confirm the Diagnosis
The indications to perform a chromosomal study are similar to the reasons to suspect a genetic syndrome and are suspected aneuploidy or other chromosomal disorder including of sex chromosomes, newborn with ambiguous genitalia, balanced translocation or family history of known rearrangement and couples with recurrent pregnancy losses. [16] In order to confirm the diagnosis in a dysmorphic neonate cytogenetics and molecular genetics investigation which are available are as follows:-
- Karyotype
- Fluorescence in situ hybridization (FISH)
- DNA microarray
- Comparative genomic hybridization (CGH)
- Single nucleotide polymorphism (SNP) or oligonucleotide arrays
- Molecular analysis
- Biochemical testing
The standard karyotype using giemsa staining (G-banding) from a peripheral blood sample can confirm a chromosomal aneuploidy including multiple malformation syndromes which result from large visible chromosome rearrangements resulting in deletion or addition of material. These rearrangements can involve an entire arm of a chromosome or may be submicroscopic. Such submicroscopic deletions can often be detected by targeted fluorescence in situ hybridization (FISH) probes specific for the deleted region, allowing the identification of the microdeletion syndromes e.g. Prader-Willi syndrome, Angelman syndrome, Smith-Magenis syndrome, Miller-Dieker and DiGeorge syndrome. Three techniques of FISH are commonly applied in dysmorphology. First, probes that are specific to the gene locus on the chromosome, bind to the segment of DNA which is not visible by G-banding on light microscopy. If the gene is deleted the FISH probe will not bind, thus demonstrating the microdeletion. Second, in whole chromosome painting (WCP), FISH probes are specific to a complete individual chromosome, rather than a single locus, and will paint the entire chromosome. Different approaches include combining fluorescent dyes to give each chromosome pair plus the X and Y chromosomes a different colour on a single metaphase spread and examining each chromosome separately, or a restricted group of chromosomes, in individual wells on a single slide. WCP is useful for identifying the origin of additional chromosome material that is microscopically visible but not distinctive enough to be assigned to a specific chromosome. It can also be used to search for light microscopically invisible (cryptic) translocations where suspicion of a chromosome abnormality remains, despite a normal standard karyotype. The exchange of similarly sized and banded material between two chromosomes which is not visible in a standard black and white G-banded study, becomes visible because of the exchange of different colours.The third approach involves use of FISH probes specific to the subtelomeric region of the short and long arm of each chromosome.The telomeres are the ends of the chromosomes and the subtelomeric regions of chromosomes are gene rich, subject to frequent breaks and exchanges, and are difficult to visualize on a standard G-banded karyotype. Subtelomeric probes are superior to WCP techniques for uncovering cryptic translocations. Genomic variations which lie between the easily identifiable chromosomal anomalies on the standard karyotype and the single nucleotide polymorphisms (SNP) are identified using DNA microarrays. DNA microarrays detect copy number variations (CNV). Larger chromosomal rearrangements such as balanced translocations or inversions are not detectable using this method.Microarray testing can be performed in either a targeted or genome-wide fashion. Two types of DNA microarray platforms used currently are array comparative genomic hybridization (array CGH) and single nucleotide polymorphism arrays (SNP arrays). Array CGH uses bacterial artificial chromosomes containing large DNA segments as probes, or small oligonucleotides as DNA probes. SNP arrays use probes based on known polymorphisms in the human genome. SNP arrays detect gains or losses of shorter stretches of the genome as the probes for a given region are densely arrayed, and thus the sensitivity for detecting alterations of that region is higher. SNP arrays can detect consanguinity. With a single test, microarrays can detect genomic errors associated with disorders that are usually identified by cytogenetic analysis and multiple FISH studies. Microarray analysis thus provides robust and exceptional level of resolution from a diagnostic perspective. However the interpretation of the results in assigning causality and clinical significance of the multiple alterations that are detected in each individual is not clear. Towards this end, the availability of databases with information on normal variation in multiple ethnic populations and testing of unaffected parents remain standard approaches to discerning whether a CNV is responsible for the dysmorphism or likely to cause disease in the future. Based on current evidence it is recommended that Chromosomal microarray be ordered as the first tier genetic test in place of a karyotype for patients with unexplained multiple congenital anomalies. High detection rates of CNV’s in neonates with birth anomalies has been demonstrated using chromosomal microarray. Molecular testing provides several answers – confirming the diagnosis, the cause of the anomaly, identifying family members at risk.It establishes a diagnosis in a symptomatic individual, identifies the inheritance pattern, and provides carrier testing and prenatal testing. Over 2500 tests are available. Molecular testing may be for a single gene or multi-gene testing. In single gene study sequence analysis for missense, nonsense and splice site mutations is done. Small intragenic deletions/insertions are identified and testing for deletion/duplication of exons or whole gene is also done. In the ‘phenotype first approach’ clinician uses detailed phenotypic features to determine genes most likely to be mutated. In multi‐gene panels the clinician identifies a broad phenotype and a panel of genes are then tested. The panel varies depending upon the mutations prevalent in that ethnic or geographic population. Next generation sequencing tests such as the whole genome sequencing (WGS) and the whole exome sequencing (WES) are poised to revolutionize the identification and diagnosis of genetic disorders. Lastly biochemical testing for disorders like peroxisomal disorders, Smith-Lemli-Opitz syndrome and mucopolysaccharidosis is also resorted to. In addition use of tandem mass spectrometry (MS/MS) in babies suspected to have an inborn error of metabolism and neuroimaging using magnetic resonance imaging (MRI) and spectroscopy (MRS) is also often employed. [1,2,7,15,] [26-28].
Syndromes Encountered Commonly in Neonates
A list of common dysmorphic syndromes encountered during the neonatal period and the lab test for confirming the diagnosis is as given in Table 5. [16, 29]
|
Condition
|
Presenting feature
|
Diagnostic test
|
|
Trisomy 21
Down syndrome
|
Brachycephaly, simple ears, hypotonia, AVSD, single palmar crease, sandal gap,
Hirschsprung disease
|
Karyotyping
|
|
Trisomy 18
Edward syndrome
|
Contracture of fingers, globular head, dysplastic ears, low birth weight, heart defects, short great toes, radial aplasia
|
Karyotyping
|
|
Trisomy 13
Patau syndrome
|
Holoprosencephaly, cleft , heart defect, polydactyly, renal abnormalities, microphthalmia
|
Karyotyping
|
|
4p-
Wolf-Hirshorn syndrome
|
Hypertelorism, prominent glabella (Greek helmet), cleft lip and palate, short philtrum, large ears
|
May be visible on standard karyotype. More reliably detected by FISH or MLPA
|
|
5p-
Cri du Chat syndrome
|
Mewing cry, microcephaly, round face, prominent epicanthic folds, cleft palate, ear anomalies
|
Usually visible on routine karyotype. FISH will detect smaller deletions.
|
|
12p tetrasomy
Pallister Killian syndrome
|
High birth weight, macrocephaly,
diaphragmatic hernia, coarse face, hypotonia, long philtrum,
sparse hair over temples
|
Always in mosaic form.
Unlikely to be detectable on blood chromosome analysis. Need skin biopsy or FISH cells from buccal mucosa
|
|
22q11 deletion
DiGeorge syndrome Velocardiofacial syndrome
|
Cardiac defects especially outflow tract, Cleft palate, micrognathia, prominent nose,
overturned helix of ear, hypocalcaemia, absent thymus
|
FISH for 22q11 deletion
Few have smaller deletions
detectable on MLPA of
22q11
|
|
Prader-Willi syndrome
|
Neonatal hypotonia
Bitemporal narrowing,
Almond-shaped eyes,
Tube feeding required
|
DNA for 15q11-13 methylation
(15q11-13 FISH will miss
infants with uniparental
disomy (UPD) of chromosome
15)
|
|
Myotonic dystrophy
|
Hypotonia , Tented upper lip,
Elevated diaphragm
Mother has myotonia
|
Examine mother
DNA for expansion in
myotonic dystrophy gene
on chromosome 19
|
|
Beckwith- Weidemann syndrome
|
Exomphalos, High birth weight,
Large tongue, Facial naevusflammeus
|
DNA to assess methylation of 11p15
Parental DNA for UPD studies.
Not all have 11p abnormality
|
|
Cornelia De Lange syndrome
|
Low birth weight, Synophrys, hirsutism, Beaked philtrum,
Heart defects,
Limb defects but may be subtle Diaphragmatic hernia
|
Primarily a clinical diagnosis.
Some have mutations in NIPBL gene on chromosome 5 or other genes. Genetic
abnormality not found in
every patient
|
|
Neonatal Marfan syndrome
|
Long limbs, arachnodactyly,
contractures, enophthalmos, dislocated lenses, wrinkly skin, heart murmur
|
Cardiac echo and follow-up as aortic dilatation may not be present at birth. Eye
examination, FBN1 mutation analysis
|
|
Rubinstein-Taybi syndrome
|
Broad medially deviated thumbs and great toes, long columella, hirsutism, microcephaly, heart defects, glaucoma
|
Clinical diagnosis
FISH 16p13 deletion in 15-20%
Some have mutations in
CRBBP gene. Many have no
genetic abnormality identified
|
|
Goldenhar syndrome
(Hemifacialmicrosomia)
|
Findings usually unilateral.
Mandibular hypoplasia, dysplastic or absent ear, pre-auricular tags,
macrostomia, epibulbardermoid. May be vertebral and cardiac defects
|
Clinical diagnosis
Heterogeneous condition
with both genetic and
environmental causes
|
|
Achondroplasia
|
Proximal limb shortening, relatively large head, trident hand, extra skin creases, depressed nasal bridge, lumbar kyphosis
|
Skeletal survey shows square
ilia, translucent proximal
femur, narrow sacrosciatic
notch. Analysis of FGFR3 gene
shows characteristic mutation
|
|
Stickler syndrome
|
Pierre Robin sequence with cleft
palate and micrognathia. Flat nasal bridge, prominent eyes, joint laxity
|
2021 Copyright OAT. All rights reserv
Eye examination shows
myopia and vitreous
abnormalities (not often
apparent at birth). Mild
platyspondyly on spinal X-ray.
Genetic testing complex
May be mutation in Type 2 or
Type 11 collagen genes
|
Table 5: A list of common dysmorphic syndromes encountered during the neonatal period and the lab test for confirming the diagnosis.
Summary and Conclusion
The approach to diagnosis of a dysmorphic baby is similar to the approach of making a diagnosis as elsewhere in medicine. It requires a methodical approach beginning with history with emphasis on the perinatal and family history with a three generation analysis of the family tree. An examination of the antenatal maternal investigations such as the ultrasound, anomaly scans and serum triple screen results is essential. Knowledge of the common clinical signs in dysmorphic syndromes and the major and minor anomalies is required. A diagnosis ‘by gestalt’ is for the experienced clinician. Characterizing the major and minor anomalies, cross referencing of two or more anomalies and performing a syndrome search on a dysmorphology database or compendium is more likely to be fruitful.With the differential diagnosis in mind investigations such as a standard karyotype, targeted FISH study, or molecular studies need to be ordered. Based on current evidence it is recommended that chromosomal microarray should be ordered as the first tier genetic test in place of a karyotype for neonates with multiple congenital anomalies.
Key Learning Points
- The diagnosis of a dysmorphic neonate is a systematic exercise, with no recourse other than a meticulous history and a thorough and repeated clinical examination.
- One must curtail the inherent tendency to make ‘spot’ diagnosis as they can be wrong and induce anxiety in the parents. Be sensitive towards the stress the parents are facing.
- One must get familiar with the common clinical signs in dysmorphic syndromes and the minor anomalies that occur.
- Meticulous recording of growth parameters and physical measurements and comparing them with age-appropriate standards as given in the Handbook of Physical measurements, and characterizing all major and minor anomalies is crucial.
- Familiarity with dysmorphology databases and cross referencing the anomalies especially the rarer ones helps in narrowing the differential diagnosis.
- Current evidence recommends chromosomal microarray as the first tier investigation in place of karyotyping in neonates with multiple congenital anomalies.
Contribution
Vishal Vishnu Tewari (VVT) and Ritu Mehta (RM) have contributed in conception, designing of data and writing of this article. KunalTewari (KT) was responsible for revision of the manuscript for important intellectual content. VVT is responsible for overall supervision and is the guarantor of the article.
References
- Reardon W,Donnai D (2007) Dysmorphology demystified. Arch Dis Child Fetal Neonatal Ed92: F225-229. [Crossref]
- Haldeman-Englert CR, Saitta SC, Zackai EH (2012) Evaluation of the dysmorphic infant. In: Gleason CA, Devaskar SU (9th ed) Avery’s diseases of the newborn. Philadelphia: Elsevier Saunders: 186-195.
- El-Hattab AW, Sutton VR (2012) Inborn errors of metabolism. In: Cloherty JP, Eichenwald EC, Hansen AR, Stark AR (7th ed) Manual of neonatal care. India: WoltersKluver767-790.
- Jones KL, Jones MC, del Campo M (2013) In: (7th ed) Smith’s Recognisable Patterns of Human Malformations. Philadelphia: Elsevier Saunders133-134.
- Moore KL, Persaud TVN (1998) Organogenetic periods: the fourth to eight week. In: (6th ed) Developing human: clinically oriented embryology. Philadelphia: Saunders116-198.
- Sadler TW, Langman J (1990) In: (6th ed). Langmans medical embryology. Baltimore: Williams & Wilkins.
- Wynshaw-Boris A, Biesecker LG (2011) Dysmorphology. In: Kliegman RM, Stanton BF (19th ed) Nelson textbook of Pediatrics. Philadelphia: Elsevier Saunders629.
- Allanson JE,Biesecker LG, Carey JC, Hennekam RC (2009) Elements of morphology: introduction. Am J Med Genet A149A: 2-5. [Crossref]
- Allanson JE,Cunniff C, Hoyme HE, McGaughran J, Muenke M, et al. (2009) Elements of morphology: standard terminology for the head and face. Am J Med Genet A149A: 6-28. [Crossref]
- Hall BD, Graham JM Jr, Cassidy SB, Opitz JM (2009) Elements of morphology: standard terminology for the periorbital region.Am J Med Genet A 149A: 29-39. [Crossref]
- Hunter A,Frias JL, Gillessen-Kaesbach G, Hughes H, Jones KL, et al. (2009) Elements of morphology: standard terminology for the ear. Am J Med Genet A 149A: 40-60. [Crossref]
- Hennekam RC, Cormier-Daire V, Hall JG, Méhes K, Patton M, et al. (2009) Elements of morphology: standard terminology for the nose and philtrum.Am J Med Genet A 149A: 61-76. [Crossref]
- Carey JC, Cohen MM Jr, Curry CJ, Devriendt K, Holmes LB, et al. (2009) Elements of morphology: standard terminology for the lips, mouth, and oral region.Am J Med Genet A 149A: 77-92. [Crossref]
- Biesecker LG,Aase JM, Clericuzio C, Gurrieri F, Temple IK, et al. (2009) Elementsof morphology: standard terminology for the hands and feet.Am J Med Genet A 149A: 93-127. [Crossref]
- Hunter AG (2002) Medical genetics: 2. The diagnostic approach to the child with dysmorphic signs. CMAJ 167: 367-372. [Crossref]
- Kingston HM (2002) ABC of medical genetics, 3rd edn. BMJ books.
- Hall JG, Allanson JE, Gripp KW, Slavotinek AM (2007) (2nd ed) Handbook of physical measurements. New York: Oxford University press.
- Allanson JE (1997) Objective techniques for craniofacial assessment: what are the choices? Am J Med Genet 70: 1-5 [Crossref].
- Pagon RA, Adam MP, Bird TD, Dolan CR, Fong C, Stephans K (2013) University of Washington Seattle. GeneReviewsTM – NCBI Bookshelf. [Crossref]
- Bichet DG (2009) V2R mutations and nephrogenic diabetes insipidus.,ProgMolBiolTranslSci 89: 15-29. [Crossref]
- http://www.ncbi.nlm.nih.gov/pubmed
- http://www.genetests.org/
- http://www.lmdatabases.com
- http://www.possum.net.au
- Miller TD, Adam MP, Aradhya S et al. (2010) Chromosomal microarray is a first tier diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet86:749-764.
- Bosch JR, Manning MA, Cherry AM (2010) Further advances in neonatal genetics testing. Neoreviews13:e20.
- Lu XY,Phung MT, Shaw CA, Pham K, Neil SE, et al. (2008) Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics122: 1310-1318. [Crossref]
- Crotwell PL, Hoyme HE (2012) Core concepts-chromosomal aneuploidies. Neoreviews13:e30.