Abstract
Many drugs, such as sulfourea, rapid-acting insulin secretagogues, biguanides, thiazolidines, alpha-glucosidase inhibitors, sodium glucose cotransporter 2 inhibitors, and dipeptidyl peptidase-4 inhibitors, have been developed for treating diabetes orally. Currently, it is possible to choose from these drugs to specifically treat the condition of the individual patient. However, blood glucose control of most oral diabetes drugs gradually diminishes, necessitating blood glucose control by insulin. It has been indicated that the glucotoxicity and lipotoxicity of oral diabetes drugs cause malfunction of the pancreatic β cells, leading to a decrease in the pancreatic β cells by apoptosis. Oral diabetes drugs that can control blood glucose levels and protect the pancreatic β cells are under development. In this review, we will discuss the current developmental status of oral diabetes drugs and the possibility of treatments that can preserve the function of pancreatic β cells.
Key words
type 2 diabetes, incretins, GPR40, GPR119, glucokinase activators, 11β-hydroxysteroid dehydrogenase inhibitors
Introduction
According to an announcement by the International Diabetes Federation (IDF) in 2014, the number of people with diabetes is 387 million worldwide (prevalence, 8.3%) [1]. Diabetes is a chronic disease that significantly decreases the quality of life (QOL) in patients through complications such as retinopathy, neuropathy, nephropathy, and cardiovascular disorders. Not only the medical burden, but also the economic burden is huge. Patients with type 2 diabetes account for 90% of diabetes cases, and the incidence of type 2 diabetes is particularly increasing in people 40-59 years of age [1]. It is expected that appropriate blood glucose control be carried out from an early stage to prevent diabetic complications (especially cardiovascular events) that can lead to decreased QOL in patients [2]. However, the United Kingdom Prospective Diabetes Study (UKPDS) reported that, with increasing age, functional decline of the pancreatic β cells occurs; the pancreatic β cells are important for blood glucose control, and blood glucose control therefore becomes worse when their function declines [3]. In addition, a five-year follow-up survey conducted by the “A Diabetes Outcome Progression Trial” (ADOPT study) confirmed that blood glucose control by metformin or sulfourea (SUs) worsens with age, although blood glucose is well-controlled just after these drugs are first administered [4]. The age-related decrease in pancreatic β cell function is considered to be associated with lipotoxicity engendered by free fatty acids [5-7]. It is known that free fatty acids promote glucose-stimulated insulin secretion (GSIS) in pancreatic β cells via the pathway of G protein-coupled receptor 40 (GPR40) or the pathway of intracellular fatty acyl-coenzyme A (FA-CoA) [8,9] (Figure 1). However, the exposure of pancreatic β cells to highly concentrated free fatty acids over the long term increases the expression of carnitine palmitoyltransferase1 (CPT-1) and uncoupling protein-2 (UCP-2) and decreases FA-CoA levels, leading to a decrease in GSIS [5-7]. Moreover, it is considered that apoptosis of pancreatic β cells is easily induced by oxidative stress caused by glucotoxicity and oxidized low-density lipoprotein (LDL) [10,11]. It has been confirmed in diabetic patients and in a diabetic mouse model that pancreatic β cells decrease by apoptosis [12,13]. In addition, hypoglycemia is of concern because the promotion of insulin secretion by SUs does not depend on the concentration of glucose [14]. Although strict blood glucose control is important for the inhibition of cardiovascular events in diabetic patients [2], hypoglycemia increases the risk of cardiovascular events [15]. Therefore, medicines that promote GSIS or maintain pancreatic β cell function are needed.
Incretin-based drugs expected to protect pancreatic β cells
Incretins are gastrointestinal hormones secreted from the small intestine. The two main incretins are glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) (Figure 1). Approximately 10 years ago, the US Food and Drug Administration (FDA) approved exenatide, which is a GLP-1 analogue and one of several incretin-based drugs. The relationship between exenatide therapy and the risk of pancreatitis has been pointed out in case reports that were published after the clinical trial and release of the drug [16,17]. However, the increased risk for pancreatitis produced by incretin-based drugs was negated by the results of meta-analysis studies and cohort studies [18,19]. In addition to the promotion of GSIS, incretin-based drugs also protect pancreatic β cells through long-term administration [20-22].
Lipotoxicity, which causes malfunction of pancreatic β cells, can be reduced by the ATP-binding cassette, subfamily A member 1 (ABCA1) transporter, which promotes cholesterol efflux from cells. Loss of function of ABCA1 in pancreatic β cells results in the accumulation of cholesterol and a reduction in insulin secretion [23,24]. In contrast, increased expression of ABCA1 leads to improved insulin secretion and protection of pancreatic β cells from lipotoxicity [25]. Li et al. reported that exendin-4, a GLP-1 agonist, induced the expression of ABCA1 in pancreatic β cells via the CaMKK/CaMKIV signaling pathway [26]. Therefore, the induction of ABCA1 by GLP-1 is likely to protect pancreatic β cells from lipotoxicity. In addition, GLP-1 agonists induce the expression of B-cell lymphoma 2 (BCL2), an anti-apoptotic protein, and reduce the expression of caspase-3, a protein with a central role in the execution phase of apoptosis. GLP-1 agonists also inhibit apoptosis induced by glucose or fats [27,28]. Thus, it is considered that GLP-1 agonists function to protect pancreatic β cells as well as improve their insulin secretion.
Development of oral diabetes drugs
Incretin-based drugs offer superior GSIS promotion and protect pancreatic β cells, and have extensively changed the treatment of type 2 diabetes. Incretin-based drugs must be administered by injection because incretin-based drugs are GLP-1 analogues. Thus, incretin-based drugs have the disadvantage that they cannot be administered to all patients, necessitating the development of new oral diabetes drugs. Candidates for new oral diabetes drugs are G protein-coupled receptor (GPR) 40 agonists, GRP119 agonists, glucokinase activators, and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors [29-37] (Tables 1 and 2).
Table 1. The different types of oral diabetes drugs under development
Target |
Compound |
Company |
Status |
Ref. |
GPR40 agonist
GPR119 agonist
glucokinase activator
11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor
Other |
Fasiglifam (TAK-875)
JTT-851
LY2881835
PSN821
MBX-2982
GSK1292263
DS-8500a
TAK-329
PF-04937319
AZD6370
AZD1656
Piragliatin
PF-00915275
INCB-13739
Colestilan
|
Takeda
Japan Tobacco
Eli Lilly
Prosidion
CymaBay Therapeutics
GlaxoSmithKline
Daiichi-Sankyo
Takeda
Pfizer
AstraZeneca
AstraZeneca
Roche
Pfizer
Incyte
Mitsubishi Tanabe |
Phase3 discontinued
Phase2
Phase1
Phase2 inetrruption
Phase2
Phase2 discontinued
Phase2
Phase1 discontinued
Phase2
Phase1 discontinued
Phase1 discontinued
Phase2
Phase1
Phase2
Phase2 additional indication |
46
29
29
30
30
30
31
33
32
56
57
52
34
64
68 |
Table 2. Structures of diabetes drugs under development
compound |
|
Ref. |
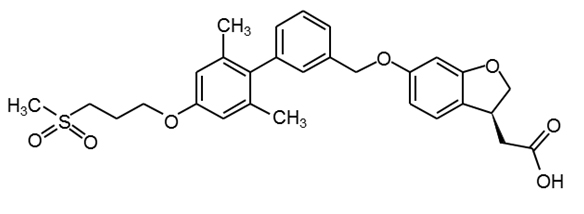
Fasiglifam
(TAK-875) |
|
29 |
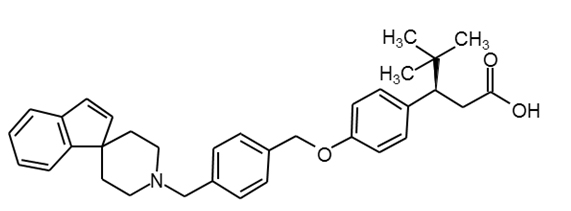
LY2881835 |
|
29 |
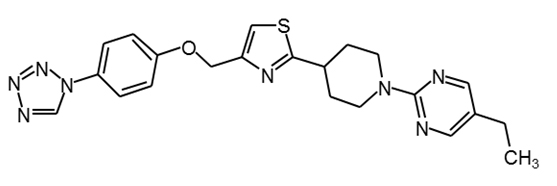
MBX-2982 |
|
30 |
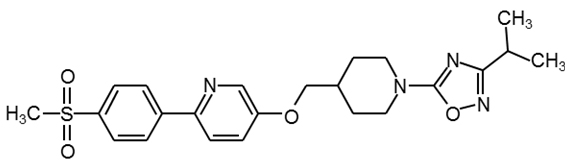
GSK1292263 |
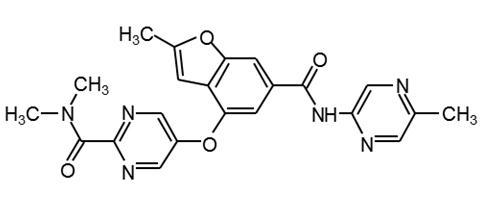
|
30 |
PF-04937319 |
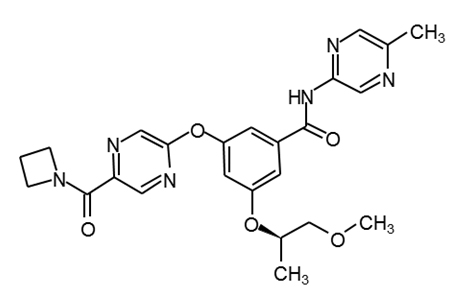
|
34 |
AZD1656 |
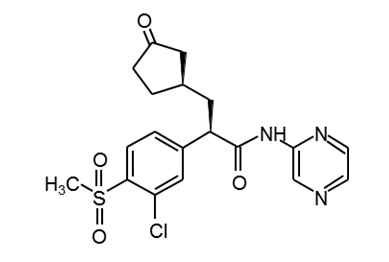
|
PubChem16039797 |
Piragliatin |
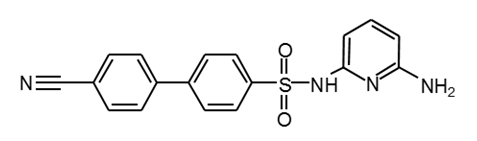
|
34 |
PF-00915275 |

|
36,37 |
Agonists for GPR40 and GPR119
GRP40 is expressed in pancreatic β cells and the intestinal tract [9,38]. GSIS is enhanced by an inositol-3-phosphate-mediated increase in the intracellular calcium concentration via GRP40 in pancreatic β cells (Figure 1) [9,39,40]. The tissue distribution of GPR40 overlaps with GPR119, a Gs-coupled receptor [41,42]. GSIS in pancreatic β cells is also enhanced by a cyclic AMP (cAMP)-mediated increase in the intracellular calcium concentration via GRP119 [42]. Moreover, both GPR40 and GPR119 are expressed in K and L cells in the small intestine, and lipids induce GLP-1 and GIP via GPR40 or GPR119 [9,29,43]. Thus, it may be possible for agonists for GPR40 and GPR119 to enhance GSIS by the direct stimulation of pancreatic β cells and induction of incretins. Moreover, since it is possible to administer agonists for GPR40 and GPR119 orally (Table 1), they are expected to be developed as alternative drugs for GLP-1 analogues that are administered by injection only. At least GPR40 agonists do not cause lipotoxicity [44,45].
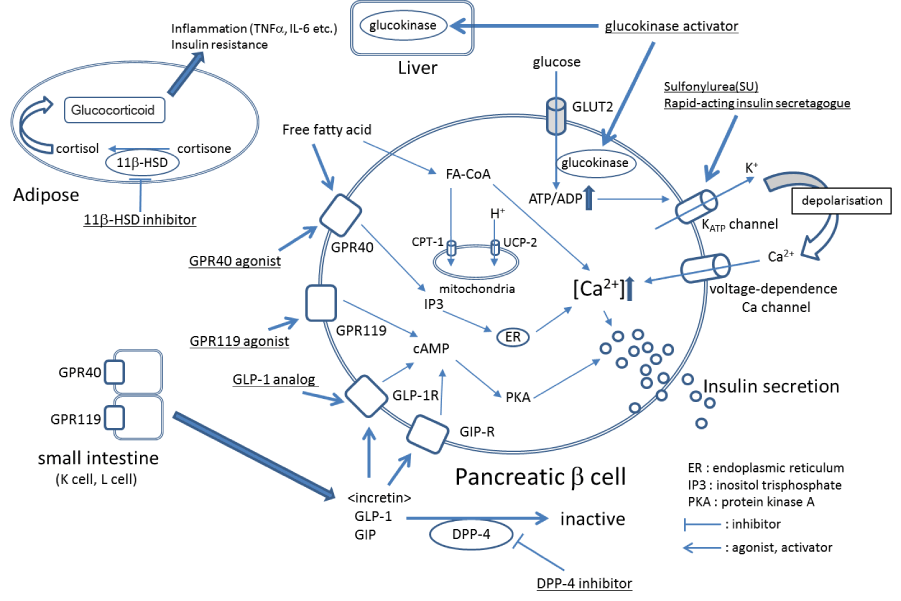
Figure 1. The mechanisms of major diabetes drugs
Fasiglifam (TAK-875) (Tables 1 and 2), the most-developed GPR40 agonist, effectively reduces blood glucose. However, the development of fasiglifam was stopped due to its hepatotoxicity [46]. There are dozens of candidate agonists for GPR40 and GPR119. JTT-851, MBX-2982, and DS-8500a (Table 2) are in phase II trials, and are expected to become new oral diabetes drugs [29-31].
Glucokinase activators
Pancreatic β cells function as glucose sensors and control GSIS. Insulin from pancreatic β cells inhibits gluconeogenesis and induces glycogen synthesis in the liver, which is the important organ in controlling blood glucose. Glucokinase is the rate-limiting enzyme that converts glucose to glucose-6-phosphate during glycolysis. The activity of glucokinase is reduced in the livers of diabetic patients [47]. Glucokinase activators, which activate glucokinase by binding to the allosteric site, are expected to promote glycometabolism in the liver and promote increased control of blood glucose [48]. In addition, it is considered that glucokinase activators contribute to the promotion of insulin secretion in pancreatic β cells via the ATP-dependent potassium channel that is opened by the glucose-dependent increase of ATP (Figure 1) [49]. Moreover, glucokinase activators stimulate the growth of pancreatic β cells and inhibit apoptosis caused by oxidative stress and glucotoxicity [50,51]. In a mouse model of diabetes, glucokinase activators have been shown to effectively reduce blood glucose and increase pancreatic β cells [49,52]. PF-04937319 and piragliatin (Tables 1 and 2) have been shown to effectively control blood glucose in clinical trials [53,54]. Although AZD1656 and AZD6370 can also effectively control blood glucose just after administration [55,56], their effectiveness in controlling blood glucose was found to decrease with long-term administration [57,58]. More clinical trials of glucokinase activators are necessary.
11 β-HSD1 inhibitors
11β-HSD1 is expressed in hepatocytes and adipocytes and converts cortisone to cortisol (Figure 1). The expression level of 11β-HSD1 is up-regulated in the adipose tissue of patients with acquired obesity. Cortisol induces insulin resistance and the secretion of inflammatory cytokines, such as tumor necrosis factor-α, interleukin (IL)-1, and IL-6, by activating the glucocorticoid receptor in adipocytes (Figure 1) [59,60]. Since a high-fat diet did not induce diabetes and dyslipidemia in 11β-HSD1–knockout mice, it is suggested that 11β-HSD1 is associated with the progression from obesity to insulin resistance and diabetes [61,62]. Metformin, a first-line drug for diabetes, together with INCB-13739 (Table 1), an 11β-HSD1 inhibitor, resulted in a 24% reduction in homeostasis model assessment-insulin resistance (HOMA-IR, the index of insulin resistance) and a 0.6% reduction in glycated hemoglobin (HbA1c) compared to metformin only [63,64]. Thus, 11β-HSD1 inhibitors can be candidate drugs for diabetes with obesity.
Other diabetes drugs under clinical trials
Many patients with type 2 diabetes also have dyslipidemia, which leads to insulin resistance and lipotoxicity in pancreatic β cells. Therefore, treatment of dyslipidemia in addition to diabetes can improve blood glucose levels. Colestilan (Table 1) is one dyslipidemia drugs that improves hypercholesterolemia and promotes the metabolism of cholesterol to bile acid through facilitating bile acid secretion. It has been known that colestilan reduces blood glucose in diabetic patients [65]. Moreover, colestilan has been observed to increase GLP-1 levels as well as reduce cholesterol levels in a mouse model of diabetes [66,67]. In a 12-week clinical trial conducted by Kondo et al., patients with type 2 diabetes received colestilan, which reduced not only LDL cholesterol levels, but also HbA1c, compared with patients with type 2 diabetes who received placebo therapy [68]. Administration of colesevelam, a dyslipidemia drug, also reduced LDL and HbA1c in patients with type 2 diabetes [69]. Colestilan is expected to become the diabetes drug of choice for treating patients with diabetes having high LDL cholesterol.
Oral diabetes drugs expected to protect pancreatic β cells
Diabetes treatment in the past has focused on the glucotoxicity created by high blood glucose levels. An important focus for diabetes treatment has been the development of drugs to control blood glucose levels by promoting insulin secretion. However, the importance of protecting pancreatic β cells and not causing hypoglycemia has been recognized.
Currently, dipeptidyl peptidase-4 (DPP-4) inhibitors, incretin-based drugs, and GLP-1 analogues are already on the market; these drugs are the most useful because they protect pancreatic β cells as well as control blood glucose. In addition, although the present GLP-1 analogues must be administered once daily, a new GLP-1 analogue has been developed for administration once weekly [70].
In this review, we described oral diabetes drugs under development that aim to protect pancreatic β cells and decrease insulin resistance. New oral diabetes drugs are expected to provide different types of treatment that can be adapted for the particular conditions of individual diabetic patients.
References
- IDF DIABETES ATLAS 6th Edition ( 2014).
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359: 1577-1589. [Crossref]
- [No authors listed] (1995) U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 44: 1249-1258. [Crossref]
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, et al. (2006) Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 355: 2427-2443. [Crossref]
2021 Copyright OAT. All rights reserv
- Oprescu AI, Bikopoulos G, Naassan A, Allister EM, Tang C, et al. (2007) Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes 56: 2927-2937. [Crossref]
- Carlsson C, Borg LA, Welsh N (1999) Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 140: 3422-3428. [Crossref]
- Dubois M, Kerr-Conte J, Gmyr V, Bouckenooghe T, Muharram G, et al. (2004) Non-esterified fatty acids are deleterious for human pancreatic islet function at physiological glucose concentration. Diabetologia 47 : 463-469 [Crossref]
- Yaney GC, Korchak HM, Corkey BE (2000) Long-chain acyl CoA regulation of protein kinase C and fatty acid potentiation of glucose-stimulated insulin secretion in clonal beta-cells. Endocrinology 141: 1989-1998. [Crossref]
- Edfalk S, Steneberg P, Edlund H (2008) Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57: 2280-2287. [Crossref]
- El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, et al. (2003) Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144: 4154-4163. [Crossref]
- Haefliger JA, Martin D, Favre D, Petremand Y, Mazzolai L, et al. (2013) Reduction of connexin36 content by ICER-1 contributes to insulin-secreting cells apoptosis induced by oxidized LDL particles. PLoS One 8: e55198. [Crossref]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, et al. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102-110. [Crossref]
- Butler AE, Janson J, Soeller WC, Butler PC. (2003) Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52 : 2304-2314. [Crossref]
- Burge MR, Sood V, Sobhy TA, Rassam AG, Schade DS (1999) Sulphonylurea-induced hypoglycaemia in type 2 diabetes mellitus: a review. Diabetes Obes Metab 1: 199-206. [Crossref]
- Goto A, Arah OA, Goto M, Terauchi Y, Noda M (2013) Severe hypoglycaemia and cardiovascular disease: systematic review and meta-analysis with bias analysis. BMJ 347: f4533. [Crossref]
- Garber A, Henry R, Ratner R, Garcia-Hernandez PA, Rodriguez-Pattzi H, et al. (2009) Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet 373 : 473-81. [Crossref]
- Denker PS, Dimarco PE (2006) Exenatide (exendin-4)-induced pancreatitis: a case report. Diabetes Care 29: 471. [Crossref]
- Li L, Shen J, Bala MM, Busse JW, Ebrahim S, et al. (2014) Incretin treatment and risk of pancreatitis in patients with type 2 diabetes mellitus: systematic review and meta-analysis of randomised and non-randomised studies. BMJ 348: g2366. [Crossref]
- Faillie JL, Azoulay L, Patenaude V, Hillaire-Buys D, Suissa S (2014) Incretin based drugs and risk of acute pancreatitis in patients with type 2 diabetes: cohort study. BMJ 348: g2780. [Crossref]
- Mari A, Sallas WM, He YL, Watson C, Ligueros-Saylan M, et al. (2005) Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed beta-cell function in patients with type 2 diabetes. J Clin Endocrinol Metab 90: 4888-4894. [Crossref]
- Gastaldelli A, Nauck MA, Balena R (2013) Eight weeks of treatment with long-acting GLP-1 analog taspoglutide improves postprandial insulin secretion and sensitivity in metformin-treated patients with type 2 diabetes. Metabolism 62: 1330-1339. [Crossref]
- Seino Y, Rasmussen MF, Zdravkovic M, Kaku K (2008) Dose-dependent improvement in glycemia with once-daily liraglutide without hypoglycemia or weight gain: A double-blind, randomized, controlled trial in Japanese patients with type 2 diabetes. Diabetes Res Clin Pract 81: 161-168. [Crossref]
- Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, et al. (2007) Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 13: 340-347. [Crossref]
- Kruit JK, Wijesekara N, Westwell-Roper C, Vanmierlo T, de Haan W, et al. (2012) Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired ß-cell function. Diabetes 61 : 659-664. [Crossref]
- Kang MH, Zhang LH, Wijesekara N, de Haan W, Butland S, et al. (2013) Regulation of ABCA1 protein expression and function in hepatic and pancreatic islet cells by miR-145. Arterioscler Thromb Vasc Biol 33: 2724-2732. [Crossref]
- Li J, Murao K, Imachi H, Masugata H, Iwama H, et al. (2010) Exendin-4 regulates pancreatic ABCA1 transcription via CaMKK/CaMKIV pathway. J Cell Mol Med 14: 1083-1087. [Crossref]
- Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, et al. (2003) Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 144: 5149-5158. [Crossref]
- Yang Y, Tong Y, Gong M, Lu Y, Wang C, et al. (2014) Activation of PPAR β/δ protects pancreatic β cells from palmitate-induced apoptosis by upregulating the expression of GLP-1 receptor. Cell Signal 26: 268-278. [Crossref]
- Defossa E, Wagner M (2014) Recent developments in the discovery of FFA1 receptor agonists as novel oral treatment for type 2 diabetes mellitus. Bioorg Med Chem Lett 24: 2991-3000. [Crossref]
- Kang SU (2013) GPR119 agonists: a promising approach for T2DM treatment? A SWOT analysis of GPR119. Drug Discov Today 18: 1309-1315. [Crossref]
- A service of the U.S. National Institutes of Health (2015) ClinicalTrials.gov Identifier: NCT02222350.
- Pal M (2009) Recent advances in glucokinase activators for the treatment of type 2 diabetes. Drug Discov Today 14: 784-792. [Crossref]
- A service of the U.S. National Institutes of Health (2012) ClinicalTrials.gov Identifier : NCT01311076.
- Pettersen JC, Litchfield J, Neef N, Schmidt SP, Shirai N, et al. (2014) The relationship of glucokinase activator-induced hypoglycemia with arteriopathy, neuronal necrosis, and peripheral neuropathy in nonclinical studies. Toxicol Pathol 42 : 696-708. [Crossref]
- Courtney R, Stewart PM, Toh M, Ndongo MN, Calle RA, et al. (2008) Modulation of 11beta-hydroxysteroid dehydrogenase (11betaHSD) activity biomarkers and pharmacokinetics of PF-00915275, a selective 11betaHSD1 inhibitor. J Clin Endocrinol Metab 93: 550-556. [Crossref]
- Thomas MP, Potter BV (2011) Crystal structures of 11β-hydroxysteroid dehydrogenase type 1 and their use in drug discovery. Future Med Chem 3: 367-390. [Crossref]
- Nair SK, Matthews JJ, Cripps SJ, Cheng H, Hoffman JE, et al. (2013) N-(Pyridin-2-yl) arylsulfonamide inhibitors of 11ß-hydroxysteroid dehydrogenase type 1: strategies to eliminate reactive metabolites. Bioorg Med Chem Lett 23 : 2344-2348. [Crossref]
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, et al. (2003) The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem 278: 11303-11311. [Crossref]
- Hara T, Hirasawa A, Sun Q, Sadakane K, Itsubo C, et al. (2009) Novel selective ligands for free fatty acid receptors GPR120 and GPR40. Naunyn Schmiedebergs Arch Pharmacol 380: 247-255. [Crossref]
- Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD (2005) Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem Biophys Res Commun 335: 97-104. [Crossref]
- Odori S, Hosoda K, Tomita T, Fujikura J, Kusakabe T, et al. (2013) GPR119 expression in normal human tissues and islet cell tumors: evidence for its islet-gastrointestinal distribution, expression in pancreatic beta and alpha cells, and involvement in islet function. Metabolism 62: 70-78. [Crossref]
- Hansen HS, Rosenkilde MM, Holst JJ, Schwartz TW (2012) GPR119 as a fat sensor. Trends Pharmacol Sci 33: 374-381. [Crossref]
- Hansen KB, Rosenkilde MM, Knop FK, Wellner N, Diep TA, et al. (2011) 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab 96: E1409-1417. [Crossref]
- Tsujihata Y, Ito R, Suzuki M, Harada A, Negoro N, et al. (2011) TAK-875, an orally available G protein-coupled receptor 40/free fatty acid receptor 1 agonist, enhances glucose-dependent insulin secretion and improves both postprandial and fasting hyperglycemia in type 2 diabetic rats. J Pharmacol Exp Ther 339 : 228-237. [Crossref]
- Wu P, Yang L, Shen X. (2010) The relationship between GPR40 and lipotoxicity of the pancreatic ß-cells as well as the effect of pioglitazone. Biochem Biophys Res Commun 403 : 36-39. [Crossref]
- Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R (2015) Efficacy and safety of fasiglifam (TAK-875), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebo-controlled, phase III trial. Diabetes Obes Metab 17: 675-681. [Crossref]
- Haeusler RA, Camastra S, Astiarraga B, Nannipieri M, Anselmino M, et al. (2014) Decreased expression of hepatic glucokinase in type 2 diabetes. Mol Metab 4: 222-226. [Crossref]
- Futamura M, Hosaka H, Kadotani A, Shimazaki H, Sasaki K, et al. (2006) An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem 281: 37668-37674. [Crossref]
- Al-Hasani H, Tschöp MH, Cushman SW (2003) Two birds with one stone: novel glucokinase activator stimulates glucose-induced pancreatic insulin secretion and augments hepatic glucose metabolism. Mol Interv 3: 367-370. [Crossref]
- Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, et al. (2011) Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 13: 440-449. [Crossref]
- Oh YS, Lee YJ, Park K, Choi HH, Yoo S, et al. (2014) Treatment with glucokinase activator, YH-GKA, increases cell proliferation and decreases glucotoxic apoptosis in INS-1 cells. Eur J Pharm Sci 51: 137-145. [Crossref]
- Futamura M, Yao J, Li X, Bergeron R, Tran JL, et al. (2012) Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat. Diabetologia 55: 1071-1080. [Crossref]
- Sarabu R, Bizzarro FT, Corbett WL, Dvorozniak MT, Geng W, et al. (2012) Discovery of piragliatin--first glucokinase activator studied in type 2 diabetic patients. J Med Chem 55: 7021-7036. [Crossref]
- Amin NB, Aggarwal N, Pall D, Paragh G, Denney WS, et al. (2015) Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes. Diabetes Obes Metab 17: 751-759. [Crossref]
- Norjavaara E, Ericsson H, Sjöberg F, Leonsson-Zachrisson M, Sjöstrand M, et al. (2012) Glucokinase activators AZD6370 and AZD1656 do not affect the central counterregulatory response to hypoglycemia in healthy males. J Clin Endocrinol Metab 97: 3319-3325. [Crossref]
- Ericsson H, Sjöberg F, Heijer M, Dorani H, Johansson P, et al. (2012) The glucokinase activator AZD6370 decreases fasting and postprandial glucose in type 2 diabetes mellitus patients with effects influenced by dosing regimen and food. Diabetes Res Clin Pract 98: 436-444.[Crossref]
- Wilding JP, Leonsson-Zachrisson M, Wessman C, Johnsson E (2013) Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab 15: 750-759. [Crossref]
- Kiyosue A, Hayashi N, Komori H, Leonsson-Zachrisson M, Johnsson E (2013) Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab 15: 923-930. [Crossref]
- Kannisto K, Pietiläinen KH, Ehrenborg E, Rissanen A, Kaprio J, et al. (2004) Overexpression of 11beta-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: studies in young adult monozygotic twins. J Clin Endocrinol Metab 89: 4414-4421. [Crossref]
- Duvnjak L, Duvnjak M (2009) The metabolic syndrome - an ongoing story. J Physiol Pharmacol 60 Suppl 7: 19-24. [Crossref]
- Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, et al. (2004) Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 53: 931-938. [Crossref]
- Morton NM, Holmes MC, Fiévet C, Staels B, Tailleux A, et al. (2001) Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 276: 41293-41300. [Crossref]
- Hollis G, Huber R (2011) 11β-Hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes Obes Metab 13: 1-6. [Crossref]
- Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, et al. (2010) The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33: 1516-1522. [Crossref]
- Garg A, Grundy SM (1994) Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med 121: 416-422. [Crossref]
- Chen L, McNulty J, Anderson D, Liu Y, Nystrom C, et al. (2010) Cholestyramine reverses hyperglycemia and enhances glucose-stimulated glucagon-like peptide 1 release in Zucker diabetic fatty rats. J Pharmacol Exp Ther 334: 164-170. [Crossref]
- Potthoff MJ, Potts A, He T, Duarte JA, et al. (2013) Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol 304: G371-380. [Crossref]
- Kondo K, Kadowaki T (2010) Colestilan monotherapy significantly improves glycaemic control and LDL cholesterol levels in patients with type 2 diabetes: a randomized double-blind placebo-controlled study. Diabetes Obes Metab 12: 246-251. [Crossref]
- Beysen C, Murphy EJ, Deines K, Chan M, Tsang E, et al. (2012) Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: a randomised controlled study. Diabetologia 55: 432-442. [Crossref]
- Dungan KM, Povedano ST, Forst T, González JG, Atisso C, et al. (2014) Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet 384:1349-1357.