Abstract
Parabens are esters of para-hydroxybenzoic acid extensively used as preservatives in cosmetics and pharmaceutics. In the recent years their safe use has been questioned, mainly due to their estrogenic activity, their concentration in breast cancer tissues and their endocrine disrupting consequences on the reproductive system. In this study, the interaction of five forms of paraben ligands (namely methyl, ethyl, propyl, butyl and heptyl) with a model dipalmitoylphosphatidylcholine bilayer and the human serum albumin transport tprotein, was assessed using extensive atomistic molecular dynamics simulations. Free energy profiles for the insertion of each ligand into the lipid bilayer were computed along an appropriate reaction coordinate. Protein-ligand interactions were evaluated through a combination of an efficient sampling of the protein-ligand hydrogen bond network and the full system side chains, together with an all-atom force field. Simulation results show that among the paraben ligands studied, methyl, propyl and butyl parabens penetrate more easily the bilayer and show higher binding affinity to human serum albumin in terms of their hydrogen bonding network, supporting the experimental hypotheses of their potential risk to human health.
Keywords
Parabens; HSA; DPPC; Molecular Dynamics; Docking
Introduction
Even though cosmetics have been used for many years, modern life’s current levels of body care products used regularly on the human body are of unknown precedent [1] and should be a subject of scientific research. It has been extensively suggested in the literature that esters of para-hydroxybenzoic acid, known as parabens, have incidence in the development of human breast cancer, affect the male reproductive system and are related with the development of malignant melanoma [2-7]. Moreover, even under the most prudent regulation on individual chemicals, low-dose long-term effects of mixtures over a life period of 80 years are yet unnoticed. Already in 2002 a study showed how man-made estrogenic chemicals have combined effects in an additive manner, leading to dramatic enhancement of steroid hormone’s action, even when each individual agent was present below its no-observedeffect- concentration [8,9]. For example, a recent study in rat pituitary GH3 cells describes how potential interactions between bisphenol A (BPA) and isobutylparaben may have additionally increased estrogenic potency via an estrogen receptor-mediated pathway [10].
Parabens function as antimicrobial agents specially against molds, yeasts and gram-positive bacteria [11]. Parabens are used as preservatives by the cosmetic industries in a wide variety of products (deodorants, body creams, shampoos and sun care products) [12,13]. Therefore, body-care products allow for repeated paraben exposure and absorption [14], in some cases through breast skin (a straightforward target fatty tissue for lipophilic and hydrophilic toxins) and into human milk [15]. Moreover, current human use of parabens is such that they can currently be detected in house dust [16]. Unfortunately, parabens used in cosmetics receive little consideration in terms of toxicity regulations [17]. Long-term dermal exposure to parabens might be of special interest because human skin epidermal extracts hydrolize parabens at much lower rates than other tissues [18]. Besides, as pointed out in several studies, reduced skin esterase activity might be considered one of the causes of skin toxicities [19-21].
In this study, we have used extensive atomistic molecular dynamics (MD) simulations to investigate the interaction of five common forms of parabens, namely methyl (MPB), ethyl (EPB), propyl (PPB), butyl (BPB) and heptylparaben (HPB), (figure 1) with a model dipalmitoylphosphatidylcholine (DPPC) lipid bilayer patch and the Human Serum Albumin (HSA) protein. Phosphatidylcholine (PC) is the most abundant phosphpolipid in the majority of the mammalian plasma membranes [22,23] and HSA is the most abundant protein in plasma contributing to almost 80% of the blood osmotic pressure [24,25]. Other computational studies have described parabens’ lipophilicity [26] and their interactions with the human oestrogen-related receptor γ [27]. However, to the best of our knowledge, free energy profiles for the insertion of each paraben ligand into the lipid bilayer are not reported in the literature and are computed here for the first time. Though, given that parabens reach the circulation system mainly by dermal application and HSA is responsible for interacting with different drugs, such as aspirin, ibuprofen, steroids and long chain fatty acids [25-30], it is of biological interest to study the underlying molecular mechanisms of these common-use molecules while penetrating the biomembrane and to evalu- ate their possible transportation within the body [31,32].
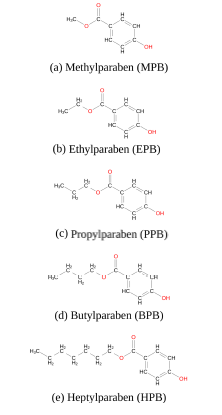
Figure 1. Five common forms of parabens
Computational methods
Lipid bilayer unrestrained simulations
To study the spontaneous penetration of paraben ligands into the bilayer, five independent systems were prepared for unrestrained atomistic MD simulations. Each system consisted in a group of 10 identical molecules of a paraben ligand, solvated in SPC [33] water box together with a 128-DPPC bilayer patch (64 lipids per leaflet), (figure 2) for MPB and supplementary material for the rest of DPPC-ligand systems). With almost 7500 water molecules (ensuring full membrane hydration) paraben molecules were initially equidistantly distributed along the center plane of the water slab (at z∼4nm from the bilayer center), keeping the bilayer normal perpendicular to the XY plane of the coordinate system. Lipids were modeled using the forcefield proposed by Berger and collaborators [34]. Systems were minimized with the steepest descent method to remove any excess of strain and potential overlaps between neighboring atoms and then equilibrated for 5ns. Production MD runs were performed for 100ns in the NPT thermodynamics ensemble using Nose- Hoover’s [35] thermostat and Parrinello- Rahman’s barostat [36] at 323K, well above the 314K phase transition temperature for DPPC [37]. Convergence was verified by a block-averaging error analysis [38] for membrane’s mean area fluctuations.
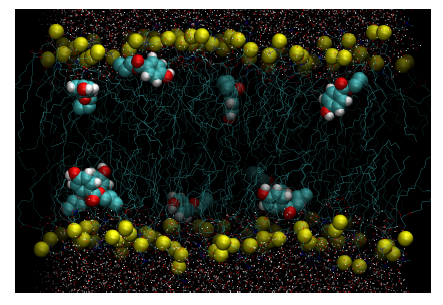
Figure 2. Configurational snapshot at 100ns extracted from the unrestrained simulation of DPPC-methylparaben. Yellow beads represent the phosphorous atoms of DPPC. Methylparaben ligands are represented with their van der Waals surfaces and water molecules by points. Equivalent figures are provided as supplementary material for the rest of DPPC-ligand systems studied.
PMF simulations
To calculate the transfer free-energy of inserting a paraben ligand into the bilayer patch, an external force was applied to the center of mass of the ligand to generate initial configurations. Therefore, umbrella sampling [39] was used to compute the potential of mean force (PMF) for the penetration of each individual paraben ligand into the bilayer. The reaction coordinate was defined as the Z-component of the distance between the center of mass of the ligand (Lcom) and the center of mass of the lipid bilayer [40-42] (Bcom), see equation 1. In this way, the ligand was free to move in the XY plane while it was pulled into the bilayer core. To span the reaction coordinate in the interval 0- 4 nm a set of 41 windows were prepared to run for 25ns in the NPT thermodynamics ensemble using Nose-Hoover’s [35] thermostat and Parrinello-Rahman’s barostat [36] at 323K. Free energy profiles were then recovered through theWeighted Histogram Analysis Method (WHAM) [43,44]. The applied harmonic potential to the reaction coordinate was set to 1000 kJmol−1nm−2. This insertion procedure is robust and it allows a gradual relaxation of the bilayer around the solute molecule [45,46]. Convergence was verified by applying WHAM on consecutive trajectory blocks of 5 ns and is provided as supplementary material.
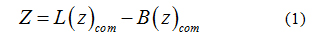
All simulations including the lipid bilayer were performed with Gromacs-4.6.3 [47] patched with Plumed 2.0.1 [48] which allows for simple implementation of reaction coordinates. In all cases a time step of 2 fs was used with all bond-lengths constrained using the sixth-order LINear Constraint Solver (LINCS) algorithm [49]. The Particle-Mesh- Ewald (PME) method was used for the long-range electrostatics [50] with reciprocal space interactions set to 0.16 nm and real space cutoffs to 1 nm. Paraben ligands were parametrized for Gromacs using the PRODGR web server [51] and the Gromos45a3 force field. The coupling time constants for the thermostat and the barostat were set to 0.5 ps and 5 ps, respectively. The reference pressure was set to 1 bar using the semi-isotropic pressure coupling type to let the bilayer to deform in the XY plane independently from the Z direction and compressibility was set to 5x10−5bar−1. Systems were subject to periodic boundary conditions in all directions.
Protein-ligand docking
To identify the most proper HSA binding site for each paraben ligand (MPB, PPB, EPB, BPB and HPB) we followed a protein-ligand docking procedure using two different approaches: (a) the freely available automatic server for molecular docking PatchDock [52] and (b) the also freely available server for Protein Energy Landscape Exploration (PELE) [53]. PatchDock is a large-scale rigid-body docking web-tool that allows for structure prediction and scoring of protein-ligand systems using shape complementarity principles and atomistic desolvation energy. PatchDock’s algorithm has three mainsteps: (i) molecular shape representation where a segmentation algorithm is applied for detection of geometric patches (concave, convex and flat surface pieces); (ii) surface patch matching through geometric hashing [54] and pose-clustering [54] techniques to evaluate the patches detected in the previous step; and (iii) filtering of all complexes with unacceptable penetrations of the atoms of the receptor to the atoms of the ligand and ranking of the remaining candidates according to a geometric shape complementarity score.
On the other hand, PELE is a heuristic algorithm that combines a Monte Carlo stochastic approach with protein structure prediction techniques. PELE is based on three main steps: (i) an initial perturbation, (ii) full system side chain sampling and (iii) a final minimization of the whole protein. PELE makes a ligand move that is accepted (defining a new minimum) or rejected based on a Metropolis criterion [55]. Flexibility in PELE is achieved by minimizing protein-ligand systems and by forcing small displacements on the alpha carbons in a low frequency anisotropic normal mode (ANM). In this way, the global motion of the receptor protein is explored (see figure 3).
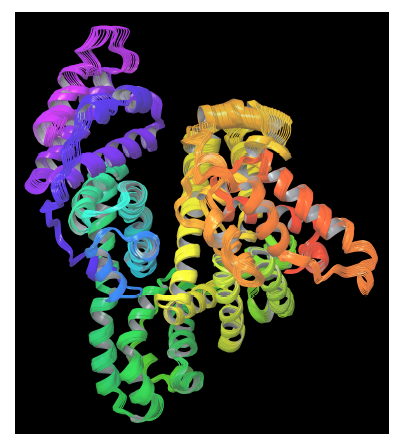
Figure 3. PELE’s protein perturbation based on a combination of low frequency normal modes obtained in an anisotropic network model (ANM). A constrained minimization forces small displacements on alpha carbons
The crystal structure of the Human Serum Albumin (HSA) is available in the Protein Data Bank (PDB ID: 1AO6) [56]. The five most common forms of paraben ligands were downloaded from PubChem [57] with codes CID7456 (MPB), CID7175 (PPB), SID125307882 (EPB), CID7184 (BPB), SID87570641 (HPB). Their 3D structures were optimized using the academic version of Maestro 9.7 [58] before docking.
Docking simulations with PatchDock provided a set of 100 candidates ranked by PatchDock’s scoring function for each of the five systems, following the standard of the Critical Assessment of Prediction of Interactions (CAPRI) for proteinprotein complexes where a near-native solution is generally found among the top 100 and very often among the top 10 solutions [59].
By using PELE to perform an unconstrained ligand exploration for binding site search, we explored the energetic landscape around the protein for 400 PELE steps, with an statistical acceptance between 30% and 40%. After completion, PELE returned the configurations corresponding to the accepted steps with their total and binding energies. Convergence was evaluated through total energies, getting in all cases variations of less than 1% between consecutive accepted steps. PELE’s binding energies were then used for scoring poses. For each protein-ligand system a top1 candidate was selected in the vicinity of the protein subdomain predicted by PatchDock with the lowest binding energy pose found by PELE. Binding and total energy evolution figures as well as PELE’s input file are provided as supplementary material.
Protein-ligand molecular dynamics pose refinement
Atomistic restrained MD simulations were also used to refine protein-ligand candidates of the five paraben molecules. By applying a harmonic restraint of 1000 kJmol−1nm−2 to the top1 configurations found by the combination of PELE and PatchDock, a collective variable D was designed to force the ligand to remain in its binding pocket, while allowing the receptor protein to freely explore its conformational space. The reaction coordinate D measured the three-dimensional distance between the center of mass of the ligand Lcom and the geometric center of the protein binding site, (Pcenter) as determined by the docking analysis for each paraben ligand (see equation 2). More details on this reaction coordinate are given in section 2.2.
All protein-ligand systems were solvated in Single Point Charge (SPC) water model [34], minimized with the steepest descent method and equilibrated for 5ns. Production MD runs where performed for 20ns under the NPT ensemble using Nose-Hoover’s [35] thermostat and Parrinello-Rahman’s barostat [36] at 298K with periodic boundary conditions in all directions. The reference pressure was set to 1 bar with the isotropic pressure coupling type, the coupling time constants for the thermostat and the barostat were set to 0.5 ps and 5 ps, respectively and compressibility was set to 4.5−10−5bar−1. The Particle-Mesh-Ewald (PME) method was used for the long-range electrostatics [50] with reciprocal space interactions spacing set to 0.16 nm and real space cutoffs to 1 nm. In all cases a time step of 2 fs was used with all bond-lengths constrained using the sixth-order LINear Constraint Solver (LINCS) algorithm [49].
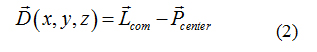
Protein, ligand and bilayer figures were created using the freely available Visual Molecular Dynamics (VMD) [60] and the academic version of Maestro 9.7 Molecular Modeling Environment [58].
Results and discussion
Ligand-membrane interactions
Lipid membranes are effective at isolating the content of the cell, yet the integrity of the lipid bilayer can be altered by a variety of external stimuli. Lipid packaging in the bilayer can be affected by any binding molecule that reaches the membrane. As extensively reported in the literature, membrane’s biophysical properties such as fluidity and phase transition temperature can be affected by external molecules that bind to the bilayer [61-63]. A downfield shift of the transition temperature has been observed experimentally for penetrating molecules mainly interacting with the phospholipid’s polar heads [64]. On the other hand, molecules intensely interacting with the phospholipid’s acyl chains, result in the lowering of the molar enthalpies (ΔH) of the main phase transition [65,66].
Unrestrained MD simulations of multiple parabens initially in bulk water allowed for the evaluation of their spontaneous adsorption into the DPPC bilayer. Simulations continued for 100ns, a sufficient amount of simulation time to converge membranes’ area in terms of a blockaveraging error analysis [38]. It was found that heptyl and ethyl parabens do not penetrate the membrane within all simulation time and instead they tend to form clusters in bulk water (see supplementary material). On the other hand, all MPB, PPB and BPB reach the bilayer surface at different rates and do not leave the membrane again (Table 1 and figure 4). DPPCbutylparaben simulation was extended for a total of 200ns, since at 100ns one BPB molecule was still in bulk water while the other 9 were already in the bilayer, full adsorption occurred at 104ns. Overall, unrestrained MD showed that in three out of the five cases studied, ligands spontaneously bind to the bilayer surface, (figure 4). To calculate the thermodynamic work required to transport each paraben ligand from the bulk of the solvent to the surface and to the center of the bilayer, we performed umbrella sampling simulations (see section 1.2 for computational details).
Table 1. Measured quantities for 100ns unrestrained simulations of pure DPPC and DPPC-ligand for the three cases of paraben adsorption. Times tA and tB indicate respectively when the first and the last molecule penetrates the bilayer. Heat capacity Cp, compressibility modulus KA and DPPC diffusion coefficient D where computed starting at time tB
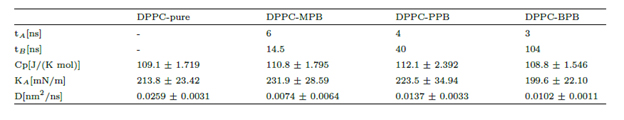
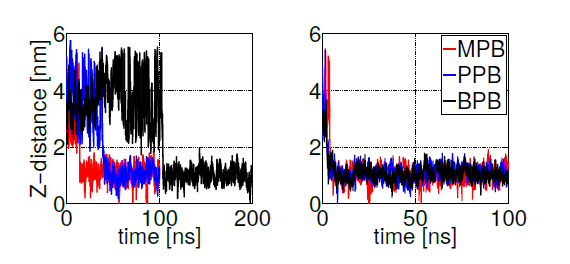
Figure 4. Z-axis projection distance between ligand and membrane center of masses. Left: last ligand to be adsorbed. Right: first ligand to be adsorbed. Heptyl and ethyl parabens were omitted since they do not bind to the bilayer within 100ns simulation time.
Free energy profiles for individual ligand insertion into the bilayer core are depicted in figure 5. As suggested by MacCallum et. al. [41], these free energy profiles in figure 5 can be divided into four regions: (1) Z∼(2.5,3.5)nm corresponds to the bulk water region with almost no population of DPPC head groups; (2) Z∼(1.5,2.5)nm includes the majority of charged phosphate and choline atoms; (3) Z∼(0.75,1.5)nm contains the final portion of the polar head groups and the initial part of the lipid tails; (4) Z∼(0,0.75)nm is composed of exclusively hydrophobic tails. Error bars in figure 5 correspond to standard errors calculated by splitting the trajectories into 10 independent blocks. Free energy minima located below the bilayer surface (at Z∼1.1nm from the center of the bilayer) for MPB, PPB and BPB with a freee energy gain bewteen 20 and 25 kJ/mol, indicate the expected position of the thermodynamic equilibrium (as suggested by unrestrained simulations). Already at Z∼2.5nm (at the begining of the bilayersurface) MPB, PPB and BPB decrease their free energies, suggesting favourable ligand-DPPC interactions in the phosphate group region. After the minimum, free energy rises up indicating unfavorable hydrophobic interactions in the lipid tails region. On the other hand, EPB and HPB show a delayed free energy decrease with respect to the other ligands while approaching to the bilayer surface, which indicates less favorable interactions with the DPPC bilayer in the phosphate region, and hence less plausible adsorption. Once inside the membrane free energy decreases till the bilayer core, showing no minima. This behavior suggests high hydrophobic interactions, specially in the lipid tails region. Overall, these free energy profiles indicate what was already partially suggested by unrestrained simulations: MPB, PPB and BPB adsorption on DPPC’s surface occurs spontaneously while their transport to the bilayer’s core is highly unfavorable. For EPB and HPB adsorption to the bilayer’s surface is less likely to happen, although once in the membrane hydrophobic interactions with lipids’ tails favourthier transport into the core.
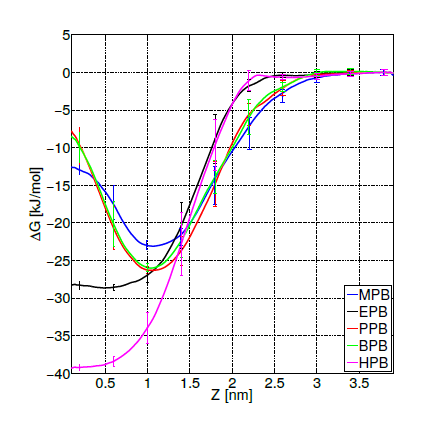
Figure 5. Free energy profiles for the insertion of methyl (MPB), ethyl (EPB), propyl (PPB), butyl (BPB) and heptyl (HPB) parabens into the bilayer core
As experimentally reported, DPPC membranes doped with propylparaben show a reduction in the transition temperature due to diminished phosphatidylcholine groups interactions, suggesting that propylparaben strongly binds and gets intercalated between the polar groups [61]. This result is in agreenment with our PMF simulations, as observed in figure 5 at Z∼2nm (phosphate groups region). To be able to compare with the other forms of parabens studied here, we calculated membranes’ biophysical parameters (such as, heat capacity, compressibility modulus and diffusion coefficients) from the previously described unrestrained simulations of DPPC bilayers doped with 10 paraben ligands (only for the cases listed in table 1, where full adsorption took place). Through a well-known statistical thermodynamics theorem [67] it is possible to convert enthalpy fluctuations (σH) into the heat capacity (Cp) by using equation 3.

Here, n is the number of atoms in the system. Analogously, to calculate the area compressibility modulus (KA) from the area fluctuations we have used equation 4. Errors in Cp and KA reported in table 1 correspond respectively to the propagation of estimates in membrane’s area and enthalpy from a block-averaging analysis [38].

To calculate DPPC diffusion coefficients for each membrane we computed the mean square displacement (MSD) for all DPPC atoms. We calculated the diffusion coefficient through a least squares fitting to the MSD. Errors listed in table 1 correspond to the difference of the diffusion coefficients from fits over the two halves of the fit interval, calculated between 10% and 90% of simulation time starting from the moment when all 10 parabens were adsorbed by the bilayer (tB).
For the three selected systems (DPPC-MPB, DPPC-PPB and DPPC-BPB), it can be observed from results summarized in table 1 that: (i) heat capacities do not change significantly beyond error bars when compare to pure DPPC, (ii) compressibilities remain as well within errors and do not show a clear dependence for these ligand concentrations and (iii) diffusion coefficients of DPPC lipids indicate that adsorbed parabens (MPB, PPB and BPB) drastically reduce lipids mobility. This result is in agreement with previous nuclear magnetic resonance (NMR) and differential scanning calorimetry (DSC) experiments for PPB indicating that this ligand strongly interacts with DPPC bilayer perturbing the membrane biophysical properties [61]. Experimental studies on dipalmitoyl phosphatidic acid (DPPA) vesicles doped with PPB indicate that PPB affects molecular mobility [68]. In figure 6 it is depicted how a PPB molecule penetrates the bilayer.
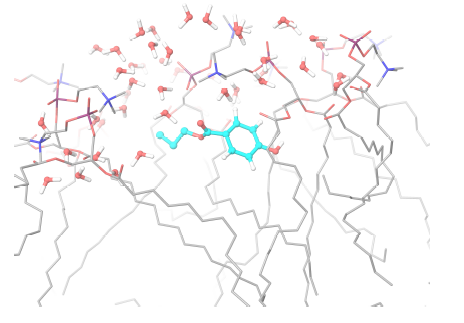
Figure 6. Propylparaben molecule (light blue) while penetrating the lipid bilayer. Only water molecules in the phospholipid’s head groups line are shown
HSA-ligand interactions
From docking calculations with PatchDock we found the binding site for all paraben ligands (except for heptylparaben) in the vicinity of HSA’a subdomain IIA (figure 7), a principal binding site together with IIIA. HSA subdomain classification is both structural and functional as domains exhibit different binding properties [69]. IIA binding site has been characterized as both electrostatic and hydrophobic [70,71] which has been also observed experimentally [24]. Correspondingly, we observed that HSA-paraben interactions are predominantly hydrophobic and polar. Heptylparaben, being the ligand with the longest chain of the ester group, remained outside from this initial binding site search due to the intrinsic approximated rigid docking approach used. Further restrained MD simulations allowed for conformational changes needed for the ligand to reach the vicinities of subdomain IIA.
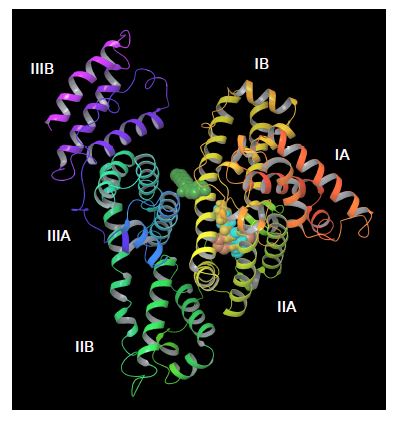
Figure 7. Docking parabens to HSA. All ligands bind in the viciniy of the IIA subdomain, except for heptylparaben (green) that remains outside due to the rigid docking first approximation
To compare the relative binding affinities between the ligands under study, we defined reaction coordinate D by measuring the three dimensional distance between the center of mass of the ligand (Lcom) and the geometric center of the protein binding site (Pcenter). Defining Pcenter as the mean between the geometrical center of the hydrophobic residues (Trp214, Leu219, Phe223, Leu234, Leu238, Leu260, Ile264, Ile290 and Ala291) of subdomain IIA [24,69] and the center of mass of thetop1 ligand pose predicted by the combination of PatchDock and PELE. By setting D=0 the collective variable keeps each paraben molecule in the pocket binding site of HSA, according to each individual docking prediction. This process combines the docking analysis information with the MD approach and forces the ligand to accommodate itself in the binding site and to explore, together with the receptor protein, its conformational space (given that by setting D=0 the restriction is applied only to the center of mass of the ligand).
These restrained MD simulations allowed for further docking refinement and, most importantly, to the evaluation of hydrogen bond formation. To do so, we simultaneously imposed a second collective variable, defined H (see equation 5), to bias dynamics into the formation of protein-ligand hydrogen bonds. The switching function form of H ensures differentiability. Accordingly, collective variable H counts the number of hydrogen bonds between a group donors and acceptors getting H = 1 if the contact between atoms i and j is formed, and H = 0 otherwise.
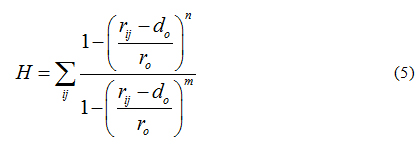
Default values were set to n=6, m=12, r0=0.3 nm and d0=0, where i counts over the group of donors (ligand) and j over the group of acceptors (protein). The harmonic restrained applied to H was set to 10 kJmol−1nm−2. Such a protocol for docking, refinement and H-bond scoring has shown to improve protein-ligand [72-74] and protein-protein [75-78] predictions.
Protein-ligand hydrogen bonding
The importance of hydrogen bonds’ spatial and directional properties have become of great interest in analyzing docking conformations [79]. Hydrogen bonds are a main contributor to the macromolecule interactions specificity, having distributions that correlate well with energy landscapes obtained through electronic structure calculations [80]. As already demonstrated, hydrogen bond refinement has shown to consistently bring initial proteinprotein models closer to natives structures [77, 78]. Currently, a wide variety of programs perform hydrogen bond optimizations combining local geometry restraints and a conformational search [81], as well as reorienting hydroxyl and thiol groups, the imidazole ring in histidines, water molecules and amide groups of ASN and GLN residues [82]. Hence, we ranked our HSA-paraben poses in terms of the amount of hydrogen bonds formed during MD trajectories under the action of collective variables D and H and we compared their relative binding affinity in these terms. Figure 8 shows protein-ligand binding affinity as measured by the sumation over 10 MD independent trajectories of reaction coordinate H (while keeping D = 0). The method gives an idea of the amount of protein-ligand hydrogen bonds generated during the complete simulation time, used here as a measure of binding affinity. In these terms, figure 8 indicates that the amount H-bonds formed by MPB, PPB and BPB parabens is systematically higher in comparison to EPB and HPB paraben forms. Noticeably, the very same ligands that spontaneously penetrate the DPPC membrane patch show more affinity to HSA in terms of their Hbond network. Such is the case of propylparaben, the most commonly used antimicrobial preservative in foods, drugs and cosmetics over 50 years [11] which recently has been suggested to induce a transformed phenotype in human breast epithelial cells in vitro [83], a possible link to breast carcinogenesis, an issue repeatedly suggested in the literature for also other forms of parabens [1-7].
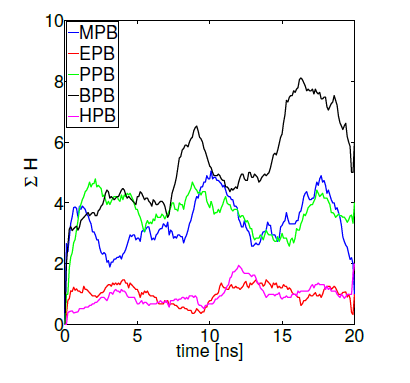
Figure 8. Parabens-HSA biding affinity as measured by reaction coordinate H
Conclusions
Molecular dynamics simulations were used to investigate the interaction of a selected group of parabens (methyl, ethyl, propyl, butyl and heptyl) with a DPPC bilayer and HSA protein. Free energy profiles for the insertion of paraben ligands into the membrane were computed by umbrella sampling, using as reaction coordinate the Z-distance between the center of masses of the bilayer and the ligand. HSA’s binding site were predicted by rigid-body protein-ligand docking and a Monte Carlo space exploration using PELE. Molecular dynamics was also used to refine protein-ligand predictions and to score them in hydrogen bonding terms. The molecular docking approach used here to locate the protein binding pocket is in agreement with the experimentally known binding site for these ligands [24,25,69]. Combining HSA results with DPPC simulations we observe that not only methyl, propyl and butyl parabens score better with HSA (figure 8) but that they also penetrate the membrane more easily (figures 4 and 5). The binding of new compounds to HSA is of major physiological relevance, as binding to HSA might be the way to control these substances concentrations as well as their side effects [32]. Moreover, the available free concentration of toxic compounds can be regulated by high binding to serum proteins [31]. Parabens have adverse implications in human health, as largely proposed by experimental studies [5,10,19]. Hence, assuming that parabens get into the circulatory system mainly through the skin, interactions of these esters of para-hydroxybenzoic acid with model lipid bilayers and the most abundant protein in human blood plasma are of major concern regarding health issues.
Acknowledgements
This work was supported by grants from CONICET (PIP 11220150100013CO01) andSeCTyP-UNCuyo. Supercomputing time provided by the Sistema Nacional de Computaci´on de Alto Desempe˜no (SNCAD-MinCyT) in clusters Mendieta (CCAD-UNC), Piluso (UNR) and Tupac (UBA) is gratefully acknowledged.
Declaration of conflicting interests
DM and FRD declared no conflicting interests with respect to the research, authorship and publication of this article.
References
- Darbre PD (2009) Underarm antiperspirants/deodorants and breast cancer. Breast Cancer Res 11 Suppl 3: S5. [crossref]
- Khanna S, Dash PR, Darbre PD (2014) Exposure to parabens at the concentration of maximal proliferative response increases migratory and invasive activity of human breast cancer cells in vitro. Journal of Applied Toxicology 34: 1051-1059.
- Charles AK, Darbre PD (2013) Combinations of parabens at concentrations measured in human breast tissue can increase proliferation of mcf-7 human breast cancer cells. Journal of Applied Toxicology 33:390-398.
- Barr L, Metaxas G, Harbach CA, Savoy LA, Darbre PD (2012) Measurement of paraben concentrations in human breast tissue at serial locations across the breast from axilla to sternum. J Appl Toxicol 32: 219-232. [crossref]
- Darbre PD, Harvey PW (2008) Paraben esters: review of recent studies of endocrine toxicity, absorption, esterase and human exposure, and discussion of potential human health risks. Journal of Applied Toxicology 28: 561-578.
- Harvey PW, Darbre P (2004) Endocrine disrupters and human health: could oestrogenic chemicals in body care cosmetics adversely affect breast cancer incidence in women? Journal of Applied Toxicology 24: 167-176.
- Harvey PW (2003) Parabens, oestrogenicity, underarm cosmetics and breast cancer: a perspective on a hypothesis. Journal of Applied Toxicology 23: 285-288.
- Rajapakse N, Silva E, Kortenkamp A (2002) Combining xenoestrogens at levels below individual noobserved- effect concentrations dramatically enhances steroid hormone action. Environ Health Perspect 110: 917-21.
- Silva E, Rajapakse N, Scholze M, Backhaus T, Ermler S, Kortenkamp A (2011). Joint effects of heterogeneous estrogenic chemicals in the e-screenexploring the applicability of concentration addition. Toxicological Sciences 122: 383-394.
- Kim SM, Jung EM, An BS, Hwang I, Vo TT, et al. (2012) Additional effects of bisphenol A and paraben on the induction of calbindin-D(9K) and progesterone receptor via an estrogen receptor pathway in rat pituitary GH3 cells. J Physiol Pharmacol 63: 445-455. [crossref]
- Soni MG, Burdock GA, Taylor SL, Greenberg NA (2001) Safety assessment of propyl paraben: a review of the published literature. Food Chem Toxicol 39: 513-532. [crossref]
- Rastogi SC1, Schouten A, de Kruijf N, Weijland JW (1995) Contents of methyl-, ethyl-, propyl-, butyl- and benzylparaben in cosmetic products. Contact Dermatitis 32: 28-30. [crossref]
- Shen HY, Jiang HL, Mao HL, Pan G, Zhou L, et al. (2007) Simultaneous determination of seven phthalates and four parabens in cosmetic products using hplc-dad and gc-ms methods. Journal of Separation Science 30: 48-54.
- Harvey PW, Darbre P (2004) Endocrine disrupters and human health: could oestrogenic chemicals in body care cosmetics adversely affect breast cancer incidence in women? Journal of Applied Toxicology 24: 167-176.
- Donovan M, Tiwary CM, Axelrod D, Sasco AJ, Jones L, et al. (2007) Personal care products that contain estrogens or xenoestrogens may increase breast cancer risk. Med Hypotheses 68: 756-766. [crossref]
- Canosa P1, Rodríguez I, Rubí E, Cela R (2007) Determination of parabens and triclosan in indoor dust using matrix solid-phase dispersion and gas chromatography with tandem mass spectrometry. Anal Chem 79: 1675-1681. [crossref]
- Harvey PW, Everett DJ (2006) Regulation of endocrine-disrupting chemicals: Critical overview and deficiencies in toxicology and risk assessment for human health. Best Practice & Research Clinical Endocrinology & Metabolism 20: 145-165.
- Lobemeier C, Tschoetsche C, Westie S, Heymann E (1996) Hydrolysis of parabenes by extracts from differing layers of human skin. Biol Chem 377: 647-651. [crossref]
- Prusakiewicz JJ, Harville HM, Zhang Y, Ackermann C, Voorman, R.L (2007) Parabens inhibit human skin estrogen sulfotransferase activity: Possible link to paraben estrogenic effects. Toxicology 232: 248-256.
- El Hussein S, Muret P, Berard M, Makki S, Humbert P (2007) Assessment of principal parabens used in cosmetics after their passage through human epidermis-dermis layers (ex-vivo study). Exp Dermatol 16: 830-836. [crossref]
- Janjua NR, Mogensen B, Andersson AM, Petersen JH, Henriksen M, et al. (2004) Systemic absorption of the sunscreens benzophenone-3, octyl-methoxycinnamate, and 3- (4-methyl-benzylidene) camphor after whole-body topical application and reproductive hormone levels in humans. J Investig Dermatol 123: 57-61.
- Ansell G, Spanner S (1982) Phosphatidylserine, phosphati-dylethanolamine and phosphatidylcholine. Biophys J. In: Hawthorne JN, Ansell GB (Eds.,)
- Rouser G, Nelson G, Fleischer S (1968) Biological Membranes. Academic Press, London.
- He XM, Carter DC (12021 Copyright OAT. All rights reservf human serum albumin. Nature 358: 209-215.
- Li L, Hitchcock AP, Cornelius R, Brash JL, Scholl A, et al. (2008) X-ray microscopy studies of protein adsorption on a phase segregated polystyrene/polymethylmethacrylate surface. 2. Effect of pH on site preference. J Phys Chem B 112: 2150-2158. [crossref]
- Casoni D, Sarbu C (2009) The lipophilicity of parabens estimated on reverse phases chemically bonded and oil-impregnated plates and calculated using different computation methods. Journal of Separation Science 32: 2377-2384.
- Zhang Z, Sun L, Hu Y, Jiao J, Hu J (2013) Inverse antagonist activities of parabens on human oestrogen-related receptor (err): In vitro and in silico studies. Toxicology and Applied Pharmacology 270: 16-22.
- Dockal M, Carter DC, Ruker F (1999) The three recombinant domains of human serum albumin: Structural characterization and ligand binding properties. Journal of Biological Chemistry 274: 29303-29310.
- Honoré B (1990) Conformational changes in human serum albumin induced by ligand binding. Pharmacol Toxicol 66 Suppl 2: 7-26. [crossref]
- Zunszain PA, Ghuman J, Komatsu T, Tsuchida E, Curry S (2003) Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct Biol 3: 6. [crossref]
- Basu A, Kumar GS2 (2014) Study on the interaction of the toxic food additive carmoisine with serum albumins: a microcalorimetric investigation. J Hazard Mater 273: 200-206. [crossref]
- Pan X, Qin P, Liu R, Wang J (2011) Characterizing the Interaction between tartrazine and two serum albumins by a hybrid spectroscopic approach. J Agric Food Chem 59: 6650-6656. [crossref]
- Berger O, Edholm O, Jähnig F (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J 72: 2002-2013. [crossref]
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration In: Pullman B (Ed) Intermolecular Forces Reidel: Dordrecht, The Netherlands, pp. 331-342.
- Nose S (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys 52: 255-268.
- Parrinello M (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 52: 7182.
- Seelig A, Seelig J (1974) Dynamic structure of fatty acyl chains in a phospholipid bilayer measured by deuterium magnetic resonance. Biochemistry 13: 4839-4845.
- Hess B (2002) Determining the shear viscosity of model liquids from molecular dynamics simulations. The Journal of Chemical Physics 116: 209-217.
- Torrie G, Valleau J (1977) Nonphysical sampling distributions in monte carlo free-energy estimation: Umbrella sampling. Journal of Computational Physics 23: 187-199.
- Marrink SJ, Berendsen HJC (1994) Simulation of water transport through a lipid membrane. The Journal of Physical Chemistry 98: 4155-4168.
- MacCallum JL, Bennett WF, Tieleman DP (2008) Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J 94: 3393-3404. [crossref]
- Porasso RD, Ale NM, Ciocco Aloia F, Masone D, Del Popolo MG, et al. (2015) Interaction of glycine, lysine, proline and histidine with dipalmitoylphosphatidylcholine lipid bilayers: a theoretical and experimental study. RSC Adv 5: 43537-43546.
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. i. the method. J. Comput. Chem 13: 1011-1021.
- Roux B (1995) The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun 91: 275-282.
- Orsi M, Essex JW (2010) Permeability of drugs and hormones through a lipid bilayer: insights from dual-resolution molecular dynamics. Soft Matter 6: 3797-3808.
- Orsi M, Sanderson WE, Essex JW (2009) Permeability of small molecules through a lipid bilayer: a multiscale simulation study. J Phys Chem B 113: 12019-12029. [crossref]
- Hess B, Kutzner C (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 4: 435-447. [crossref]
- Tribello G, Bonomi M, Branduardi D, Camilloni C, Bussi G (2014) Plumed 2: New feathers for an old bird. Computer Physics Communications 185: 604-613.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem 18: 1463-1472.
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, et al.(1995) A smooth particle mesh ewald method. J. Chem. Phys103: 8577-8593.
- Schüttelkopf AW, van Aalten DM (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 60: 1355-1363. [crossref]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33: W363-367. [crossref]
- Madadkar-Sobhani A, Guallar V (2013) PELE web server: atomistic study of biomolecular systems at your fingertips. Nucleic Acids Res 41: W322-328. [crossref]
- Wolfson HJ, Rigoutsos I (1997) Geometric hashing: An overview. IEEE Comput. Sci. Eng 4: 10-21.
- Borrelli KW, Vitalis A, Alcantara R, Guallar V (2005) PELE: Protein Energy Landscape Exploration. A Novel Monte Carlo Based Technique. J Chem Theory Comput 1: 1304-1311. [crossref]
- Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K (1999) Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng 12: 439-446. [crossref]
- Bolton EE, Wang Y, Thiessen PA, Bryant SH (2008) Chapter 12 - pubchem: Integrated platform of small molecules and biological activities, Elsevier. volume 4 of Annual Reports in Computational Chemistry, pp. 217-241.
- Schr¨odingerLLC, . Release 2014-1: Maestro, version 9.7,. Schr¨odinger, LLC, New York, NY, 2014.
- Inbar Y, Schneidman-Duhovny D, Halperin I, Oron A, Nussinov R, et al. (2005) Approaching the capri challenge with an efficient geometry-based docking. Proteins: Structure, Function, and Bioinformatics 60: 217-223
- Humphrey W, Dalke A, Schulten K (1996) VMD – Visual Molecular Dynamics. Journal of Molecular Graphics 14:33-38.
- Panicker L (2008) Interaction of propyl paraben with dipalmitoyl phosphatidylcholine bilayer: A differential scanning calorimetry and nuclear magnetic resonance study. Colloids and Surfaces B: Biointerfaces 61: 145-152.
- Jemiola-Rzeminska M, Mysliwa-Kurdziel B, Strzalka K (2002) The influence of structure and redox state of prenylquinones on thermotropic phase behaviour of phospholipids in model membranes. Chemistry and Physics of Lipids 114: 169-180.
- Gicquaud C, Auger M, Wong PT, Poyet P, Boudreau N, et al. (1996) Interaction of 4- tert-butyl-[3-(2-chloroethyl) ureido] benzene with phosphatidylcholine bilayers: A differential scanning calorimetry and infrared spectroscopy study. Archives of Biochemistry and Biophysics 334: 193-199.
- Pignatello R, Toth I, Puglisi G (2001) Structural effects of lipophilic methotrexate conjugates on model phospholipid biomembranes. Thermochimica Acta 380: 255-264.
- Castelli F, Puglisi G, Giammona G, Ventura CA (1992) Effect of the complexation of some nonsteroidal anti-inflammatory drugs with -cyclodextrin on the interaction with phosphatidylcholine liposomes. International Journal of Pharmaceutics 88: 1-8.
- Jørgensen K, Ipsen JH, Mouritsen OG, Bennett D, Zuckermann MJ (1991) A general model for the interaction of foreign molecules with lipid membranes: drugs and anaesthetics. Biochim Biophys Acta 1062: 227-238. [crossref]
- Hill T (1960) An introduction to statistical thermodynamics. Dover Publications.
- Panicker L (2007) Effect of propyl paraben on the dipalmitoyl phosphatidic acid vesicles. Journal of Colloid and Interface Science 311: 407-416.
- Artali R, Bombieri G, Calabi L, Del Pra A (2005) A molecular dynamics study of human serum albumin binding sites. Farmaco 60: 485-495. [crossref]
- Sudlow G, Birkett DJ, Wade DN (1975) The characterization of two specific drug binding sites on human serum albumin. Molecular Pharmacology 11: 824-832.
- Sudlow G, Birkett DJ, Wade DN (1976) Further characterization of specific drug binding sites on human serum albumin. Mol Pharmacol 12: 1052-1061. [crossref]
- Schneider N, Lange G, Hindle S, Klein R, Rarey M (2013) A consistent description of hydrogen bond and dehydration energies in proteinligand complexes: methods behind the hyde scoring function. Journal of Computer-Aided Molecular Design 27: 15-29.
- Wu MY, Dai DQ, Yan H (2012) Prl-dock: Protein-ligand docking based on hydrogen bond matching and probabilistic relaxation labeling. Proteins: Structure, Function, and Bioinformatics 80: 2137-2153
- Williams MA, Ladbury JE (2005) Hydrogen Bonds in Protein-Ligand Complexes. Wiley-VCH Verlag GmbH & Co. KGaA. pp. 137–161.
- Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, et al. (2003) Proteinprotein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. Journal of Molecular Biology 331: 281-299.
- Kortemme T, Morozov AV, Baker D (2003) An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and proteinprotein complexes. Journal of Molecular Biology 326: 1239-1259.
- Masone D, Vaca IC, Pons C, Recio JF, Guallar V (2012) H-bond network optimization in protein-protein complexes: are all-atom force field scores enough? Proteins 80: 818-824. [crossref]
- Masone D, Grosdidier S (2014) Collective variable driven molecular dynamics to improve proteinprotein docking scoring. Computational Biology and Chemistry 49: 1-6.
- Meyer M, Wilson P, Schomburg D (1996) Hydrogen bonding and molecular surface shape complementarity as a basis for protein docking. J Mol Biol 264: 199-210. [crossref]
- Morozov AV, Kortemme T, Tsemekhman K, Baker D (2004) Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proceedings of the National Academy of Sciences of the United States of America 101: 6946-6951.
- Bhattacharya D, Cheng J (2013) 3drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins: Structure, Function, and Bioinformatics 81: 119-131.
- Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design 27: 221-234.
- Khanna S, Darbre PD (2013) Parabens enable suspension growth of mcf-10a immortalized, nontransformed human breast epithelial cells. Journal of Applied Toxicology 33: 378-382.