Abstract
We have previously identified the common gene expression profiles among patients with non-muscle invasive and muscle invasive transitional cell carcinoma (TCC) of the urinary bladder, of both low and high grade. In the present work, we used computational tools and performed bioinformatics analysis to further investigate the presence of common Transcription Factor Binding Motifs (TFBMs) among the same TCC samples, as well as to identify the uniquely presented TFBMs in each tumor type. Gene expression data derived from four publicly available datasets were also included in our study. Cancerous samples were divided into five categories, according to their histology and metastatic potential. Our analysis revealed uniquely annotated transcription factors (TFs) with respect to each tumor category. We found that genes located in chromosomes 5, 11, 15, 17, 18 and X could be critically implicated in the progression of the disease. In particular, our data revealed that TCC of the urinary bladder implicates genes with hormone-dependent properties as well as inflammatory genes and cell cycle dependent machinery, which might play a significant role in its ontogenesis or progression. Such computational approaches could be very useful in the prediction of more targets suitable for prognostic or therapeutic purposes of the disease.
Key words
transitional cell carcinoma, urinary bladder cancer, transcription factor binding motifs, chromosomal mapping, gene ontology, linear regression
Introduction
Cancer of the urinary bladder (BC) affords the fifth most common cancer in men with peak prevalence among patients 60-70 years of age. The disease is curable if diagnosed at an early stage [1]. The majority of the tumors (~90%) arise from the transitional epithelium (TCC), whereas the remaining 10% are squamous cell carcinomas, adenocarcinomas, sarcomas, or small cell carcinomas [2]. The behavior of TCC is highly diverse and defined by two separate, but related processes: tumor recurrence and progression. At presentation, 75–85% of tumors is restricted to the mucosa, or invades the lamina propria mucosae. The remainder is either present with invasion of the muscular layer of the bladder wall or is extended to perivesical tissue, adjacent organs and the pelvic wall. More than 60% of the superficial tumors will recur at least once and progress to less differentiated or invasive neoplasms with worse prognosis in a significant percentage of patients [2]. The most useful prognostic parameters are tumor grade, stage, size, prior recurrence rate and the synchronous presence of in situ carcinoma (CIS) [3,4]. Nevertheless, a better understanding of the natural history of TCC can be expected upon the elucidation of its molecular mechanisms.
Several studies have focused on the expression profiling of BC, aiming to classify the different types, to define the biological phenotypes and to identify patterns of gene expression in superficial, muscle-invasive and metastasizing TCC [5]. Although different tumor types are expected to have differences in their expression profiles, even between individuals of the same tumor type, we hypothesized that tumors could possess similar characteristics that may eventually lead to the understanding of the aetiologies underlying carcinogenesis. Chromosome mapping is a promising method for the identification of patterns among genes, including TFBMs. The main idea is to map genes on the chromosomal regions and if correlations between them exist, they will appear through the location of genes on the chromosomal regions, since consecutive genes are often similarly expressed [6]. Therefore, we searched for common TFBMs in the promoter regions of genes that were co-deregulated in non-muscle invasive (T1) and muscle invasive (T2-T4) transitional cell carcinomas of low and high grades.
Materials and methods
Biological samples, microarray data and publicly available datasets
Our analysis included ten transitional cell carcinomas of the urinary bladder with different histology (T1-grade 1/2, T1/2-grade 3, T3-grade 3), five normal urothelium samples (controls) and the respective microarray data, as previously reported in detail [7-9]. The experimental setup was analyzed based on the reference-design [10]. All tumor samples were compared against the mean value of the controls. All microarray data are MIAME compliant and were deposited to the GEO microarray database (GSE27448). Four publicly available microarray datasets were also included in our analysis: 1) GSE89 dataset (GDS183) (40 BCs); 2) GSE3167 dataset (GDS1479) [5] (51 BCs and 9 controls); 3) GSE7476 dataset [11] (9 BCs and 3 controls) and 4) GSE12630 dataset [12] (19 BCs). In total, our pooled microarray analysis composed of 129 BC samples and 17 controls [8,9].
Data processing and computational analysis
Data processing, normalization, filtering and identification of differentially expressed (DE) genes were described in our previous reports [7-9]. We divided our data into 5 categories. The first included DE genes in all samples vs. the controls. The second, third and fourth categories were labeled as Groups “A”, “B” and “C”. In particular, Group A included T1-Grade II muscle non-invasive samples, Group B included T1-Grade III muscle non-invasive samples, Group C included T2-3-Grade III muscle invasive samples. The fifth category was composed of metastatic, high grade TCCs. A brief representation of the bioinformatics analyses is presented in Supplementary Figure 1.
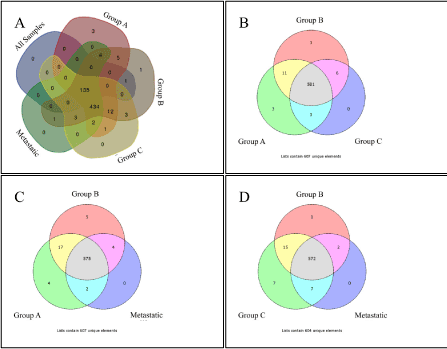
Figure 1. Venn diagrams of TF enrichment of investigated groups.
TFBM analysis in the promoters of co-deregulated genes
We investigated the TFBMs in the Transcription Element Listening System Database (TELiS) (www.telis.ucla.edu) [12], considering as promoters strings of 1000 nucleotides upstream and 200 nucleotides downstream of each gene's transcription start site (TSS) as indicated in the NCBI RefSeq database. TELiS currently contains data on 34,622 human genes. TFBMs were defined by 108 position-specific weight matrices from the JASPAR 2 database or 192 matrices representing all vertebrate TFs in the TRANSFAC database. Binding motifs were detected by the MatInspector algorithm [13]. TFBM analysis was also performed using the WebGestalt web-tool [14,15].
Chromosome mapping and linear correlations
For chromosome mapping analyses, we used the Gene Ontology Tree Machine, the WebGestalt web-tool (http://bioinfo.vanderbilt.edu/webgestalt) [15] and the Matlab® (The Mathworks Inc.) computing environment. Linear correlations were calculated using Pearson’s correlation coefficient. Values with R2 > 0.8 and p < 0.05 were statistically significant.
Gene Ontology (GO) analysis
GO analysis was initially performed using the eGOn online tool for Gene Ontology (former: http://www.genetools.microarray.ntnu.no/egon/[1]) in order to find missing gene symbols [16]. The ontology covered three domains: 1) Cellular component, the parts of a cell or its extracellular environment; 2) Molecular function, the elemental activities of a gene product at the molecular level, such as binding or catalysis; 3) Biological process, operations or sets of molecular events with a defined beginning and end, pertinent to the functioning of integrated living units: cells, tissues, organs, and organisms. The WebGestalt web-tool (http://bioinfo.vanderbilt.edu/webgestalt) [14,15] was used for the classification of gene functions. Relations of the differentially expressed genes and the TFBMs were further investigated using the Pubgene Ontology Database (www.pubgene.org). Definitions and functions of genes and TFs were based on the National Institute of Health databases (http://www.ncbi.nlm.nih.gov/gene/).
Venn diagrams
Venn diagrams were designed with the Venn web-tool from the University of Gent, Bioinformatics and Evolutionary Genomics (http://bioinformatics.psb.ugent.be/webtools/Venn/) and the web-tool Venn Diagram Generator from the Public Research Centre for Health (http://www.bioinformatics.lu/).
Results
We initially investigated the TFBM prevalence in each group. In the group entitled “all samples” we found 137 TFBMs; whereas Groups A, B and C contained 599, 602 and 591 TFBMs, respectively. The metastatic group included 582 TFBMs (p < 0.01).
GO and TFBM analysis of groups
The main biological process of the deregulated genes in each group included cellular metabolic processes and cellular component organization and biogenesis (Supplementary Figure 2). TFBM analysis also manifested various TF binding sites and therefore, potential TFs that might participate in the regulation of gene expression. Since the investigated groups were composed of BC samples of different histology, we investigated the unique TF binding sites in each group. When examining the samples in groups of three (3 different combinations) we identified the unique binding sites of the following TFs: PAX8, PPARG, STAT3, AHRARNT, AR, MYOGNF and ROAZ (Group A) and AHRARNT, EVI, ROAZ, AR and MYOGNF (Group B). No unique TF was found in Group C or in the metastatic group. Searching for unique TF binding sites among all groups, our analysis spotted PPARG, STAT3 and PAX8 in Group A and ROAZ and AHRARNT in Group B, whereas in “all samples” Group, Group C and metastatic Group no unique TF binding sites were identified (Table 1, Figure 1).
|
IN GROUPS OF THREE
|
|
Group A, Group B, Group C
|
|
UNIQUE GroupA
|
UNIQUE GroupB
|
UNIQUE GroupC
|
|
hsa_V$PAX8_01 (PAX8: Paired Box 8)
|
hsa_V$AHRARNT_02 (AHR/ARNT heterodimer: aryl hydrocarbon receptor (AHR)/ aryl hydrocarbon receptor nuclear translocator (ARNT))
|
None
|
|
hsa_V$PPARG_01 (PPARG: peroxisome proliferator activated receptor gamma)
|
hsa_V$EVI1_01 (EVI-1 also known as RUNX1: runt related transcription factor 1)
|
|
|
hsa_V$STAT3_01 (STAT3: signal transducer and activator of transcription 3)
|
hsa_V$ROAZ_01 (ROAZ also known as ZNF423: zinc finger protein 423)
|
|
|
Group A, Group B, Metastatic
|
|
UNIQUE GroupA
|
UNIQUE GroupB
|
UNIQUE Metastatic
|
|
hsa_TGTYNNNNNRGCARM_UNKNOWN
|
hsa_CRGAARNNNNCGA_UNKNOWN
|
None
|
|
hsa_V$PAX8_01 (PAX8: Paired Box 8)
|
hsa_GGCNRNWCTTYS_UNKNOWN
|
|
|
hsa_V$PPARG_01 (PPARG: peroxisome proliferator activated receptor gamma)
|
hsa_V$AHRARNT_02 (AHR/ARNT heterodimer: aryl hydrocarbon receptor (AHR)/ aryl hydrocarbon receptor nuclear translocator (ARNT))
|
|
|
hsa_V$STAT3_01 (STAT3: signal transducer and activator of transcription 3)
|
hsa_V$ROAZ_01 (ROAZ also known as ZNF423: zinc finger protein 423)
|
|
| |
hsa_YGTCCTTGR_UNKNOWN
|
|
|
Group B, Group C, Metastatic
|
|
UNIQUE GroupB
|
UNIQUE GroupC
|
UNIQUE Metastatic
|
|
hsa_GCGSCMNTTT_UNKNOWN
|
hsa_TGTYNNNNNRGCARM_UNKNOWN
|
None
|
|
hsa_V$AHRARNT_02 (AHR/ARNT heterodimer: aryl hydrocarbon receptor (AHR)/ aryl hydrocarbon receptor nuclear translocator (ARNT))
|
|
|
|
hsa_V$AR_03 (AR: Androgen Receptor)
|
|
|
|
hsa_V$MYOGNF1_01 (MYOG/NF1 heterodimer Myogenin (MYOG)/Nuclear Factor 1 (NF1))
|
|
|
|
hsa_V$ROAZ_01 (ROAZ also known as ZNF423: zinc finger protein 423)
|
|
|
|
hsa_WCTCNATGGY_UNKNOWN
|
|
|
|
hsa_YAATNANRNNNCAG_UNKNOWN
|
|
|
|
ALL FIVE GROUPS
|
|
Unique All Samples
|
Unique Group A
|
Unique Group B
|
Unique Group C
|
Unique Metastatic
|
|
None
|
hsa_V$PPARG_01 (PPARG: peroxisome proliferator activated receptor gamma)
|
hsa_V$ROAZ_01 (ROAZ also known as ZNF423: zinc finger protein 423)
|
None
|
None
|
| |
hsa_V$STAT3_01 (STAT3: signal transducer and activator of transcription 3)
|
hsa_V$AHRARNT_02 (AHR/ARNT heterodimer: aryl hydrocarbon receptor (AHR)/ aryl hydrocarbon receptor nuclear translocator (ARNT))
|
|
|
| |
hsa_V$PAX8_01 (PAX8: Paired Box 8)
|
|
|
|
Table 1. Unique TFs in BC Groups as revealed by Venn Diagram Analysis. “Group A, Group B, Group C” signifies the unique TFs found for each group as depicted in Figure 2B, similarly where it states “Group A, Group B, Metastatic” it signifies the unique TFs found for each group as depicted in Figure 2C, similarly where it states “Group B, Group C, Metastatic” it signifies the unique TFs found for each group as depicted in Figure 2D. Finally, where it states “All Five Groups” it signifies the unique TFs found for each group as depicted in Figure 2A.
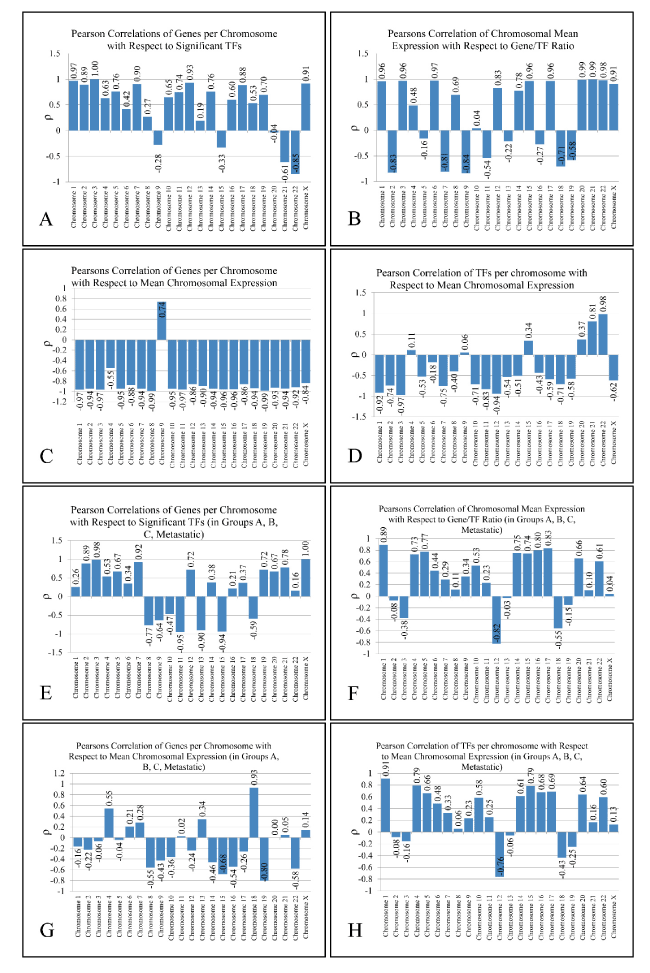
Figure 2. Metrics of Pearson’s correlation analysis. Two analyses are presented: one including all five groups that is All samples, Group A, Group B, Group B and Metastatic (A-D) and one including four groups, that is Group A, Group B, Group C and Metastatic (E-H).
Chromosomal-based GO and TFBM analysis of groups
In order to expand our search for TFBM patterns, we separated genes with respect to their chromosomal position. Especially for genes in the “all samples” Group, we performed a chromosome-specific GO analysis (Supplementary Figure 3). As expected, DE genes in most chromosomes had different GO annotations from the complete dataset, apart from those of chromosomes 1 and 6 which manifested similar profiles with the complete dataset (Supplementary Figure 2). Interestingly, functional annotation of genes on chromosomes 14 and 16 manifested cell cycle-related regulations, and on chromosome 20 the JAK-STAT cascade. Additionally, Venn diagram analysis showed the presence of numerous uniquely appearing TFBMs in each chromosome (Supplementary Figure 4). Of interest, androgen receptor (AR) appeared to be annotated on chromosome 15 only, whose genes participate in the development of muscle cells (Supplementary Figure 3). Furthermore, the NFκB binding site was appeared as unique TF binding site in chromosomes 1 (Group A) and 17 (Group C and metastatic group). Similarly, the glucocorticoid receptor (GR) appeared in chromosomes 1 (“all samples” Group) and 18 (Group C).
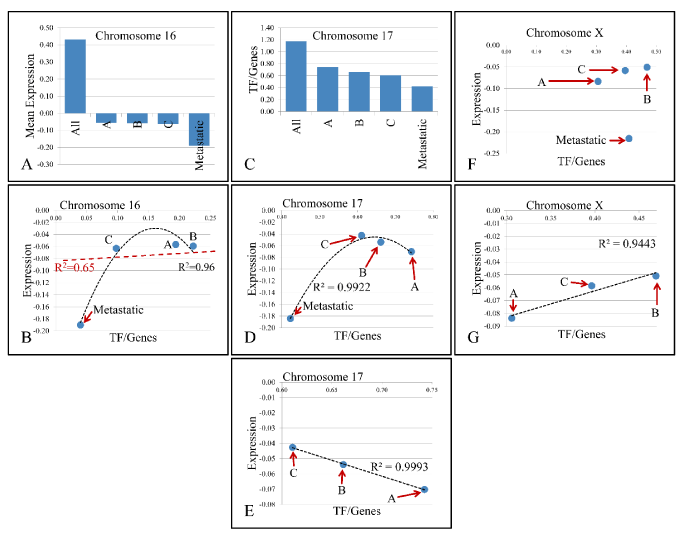
Figure 3. Descending pattern of variable values with respect to chromosomes. In particular, chromosome 16 manifested descending pattern of variation in mean gene expression as we move from All samples to Metastatic (A). Further on, regression of TF/Genes ratio vs. mean chromosomal expression levels did not manifest significant relations (B). Additionally, chromosome 17 manifested descending pattern of change in the TF/Genes ratio (C), while it presented an interesting behavior with respect to metastatic samples (D), and when analysis included only the first three groups, a significant linear relation was presented (E). Similar behavior was manifested by chromosome X (F), where the three groups except metastatic samples manifested linear dynamics (G). X-axes (in A and C) as well as abbreviations within charts are annotated as follows; All: All Samples, A: Group A- T1-Grade II muscle non-invasive samples, B: Group B- T1-Grade III muscle non-invasive samples, C: Group C- T2-3-Grade III muscle invasive samples, Metastatic: metastatic, high grade TCCs.
Pearson’s correlations of chromosomal values
Based on the number of TF binding sites found in each chromosome, we further identified their correlation with the number of the DE genes in the corresponding chromosomes. Thus, we investigated whether the activity of a chromosome and the respective TF binding sites are proportional to the number of the DE genes or the expression levels of the respective chromosome. To achieve this, we calculated four variables in each chromosome and each group: 1) the total number of DE genes (“Genes”), 2) the total number of TF binding sites in each chromosome (as revealed by the TFBM analysis) (“TFs”), 3) the ratio of binding sites of each TF versus Genes (“TFs/Genes”) and 4) the mean expression of each chromosome and each group (“Expr”) (Supplementary Figure 5).Remarkably, the “TFs/Gene” ratio and “TFs” were positively correlated (ρ > 0.9 and p < 0.05) in almost all chromosomes (apart from chromosomes 1, 12 and 17). Additionally, in chromosomes 3 and X significant correlations were observed between “TFs” vs. “Genes” (chromosome 3 and X) and “TF/Genes” vs. “Genes” (chromosome X). Two types of analyses were performed: In the first, we included all five groups, while in the second; we included Groups A, B, C and the Metastatic group. The basic metrics of our analysis are presented in Figure 2. The results of the first analysis were excluded, since the “All Samples” group was composed of genes that were simultaneously deregulated in all tumor samples, thus adding a bias to our Pearson’s correlation analysis. Our results drove us towards another aspect of chromosomal-based analysis: the presence of linear relations detected among TFs and annotated genes in each chromosome.
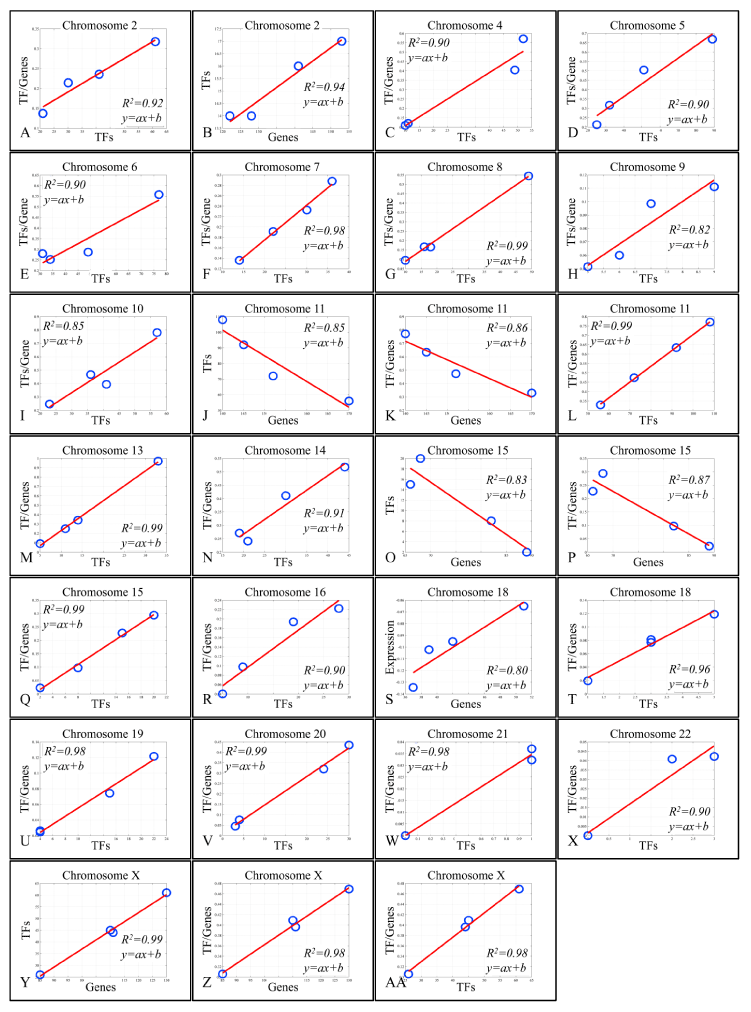
Figure 4. Regression analysis of chromosomal-based variables
Regression analysis of chromosomal variables
The presence of linear correlations among chromosomal values, gave us a further hint on the probable relations between gene expression and the annotated TF binding sites. For this purpose, we performed linear regression analyses on the calculated values. Looking for linear patterns within our data we found that chromosomes 16 and 17 manifested a descending order of chromosomal values. In chromosome 16, we found that the chromosomal mean expression decreased, moving from the “All Samples” group to the Metastatic group. Likewise, in chromosome 17, the ratio of “TF/Genes” descended as moved from “All Samples” group to Metastatic group. This finding indicated a key role of these chromosomes with respect to tumor properties. A very interesting behavior was observed in chromosome 17. While the regression in chromosome 16 between mean chromosomal expression levels and the “TF/genes” ratio did not manifest significant correlations, in chromosome 17, regression of the four groups revealed that metastatic samples were separated from the rest three groups. When Groups A, B and C were analyzed together, a significant linear relation emerged with a descending order from Group C (more aggressive tumor type) to Group A (less aggressive tumor type). A similar behavior was presented in chromosome X, where the regression between chromosomal mean expression values and the “TF/Genes” ratio did not manifest a significant pattern, while regression of the three groups (excluding the metastatic group) manifested a linear correlation (Figure 3). Regression analysis also showed that several variables manifested linear behavior in each chromosome. Most of the chromosomes manifested linear behavior with respect to “TFs” and “TF/Genes” variables. Additional linear behavior was manifested in chromosome 2 with respect to “TFs” and “Genes”, in chromosome 11 with respect to “TFs” and “Genes”, with respect to “TF/Genes” and “Genes”, in chromosome 15 with respect to “TFs” and “Genes”, in chromosome 18 with respect to “TFs” and “Genes” and chromosome X with respect to “TFs” and “Genes” and with respect to “TF/Genes” and “Genes” (Figure 4).
Discussion
In the present study, we detected the common TFBMs mapped on the promoter regions of the co-deregulated genes among different BC subtypes and investigated the presence of linear correlations between gene expression and the annotated TF binding sites.
Unique transcription factor binding motifs identified in each group
Peroxisome proliferator-activated receptor-γ (PPAR-γ) was among the uniquely annotated TFs based on the binding sites identified in tumors of Group A (T1, grade II). PPARγ is a nuclear hormone receptor and a ligand-activated TF important for urothelial differentiation. Its expression has been associated with the differentiation of the presumptive urothelium of the mouse urogenital sinus and the mature urothelium of mice, rabbits and humans [17]. PPARγ agonists, such as troglitazone (TZ), in combination with EGFR inhibition have been reported to activate the urothelial differentiation of cells in vitro [17] and are being used in the clinical setting as potential therapies [18].
STAT3 (Signal transducer and activator of transcription 3) was also uniquely annotated in Group A. This TF is a member of the STAT protein family being activated through phosphorylation in response to various cytokines and growth factors (IFNs, EGF, IL5, IL6, HGF, LIF and BMP2). It plays a key role in cell growth, apoptosis, tumor progression and proliferation [19]. Furthermore, STAT3 silencing has been connected to the suppression of cell proliferation and survival [20].
Similarly, among Group B tumors (T1-grade III), ROAZ (or ZNF423) and AHRARNT were the uniquely annotated TFs based on the binding sites that we identified. To our knowledge, there are no known reports linking ROAZ with TCC of the urinary bladder. ARNT encodes the aryl hydrocarbon receptor nuclear translocator protein that forms a complex with ligand-bound aryl hydrocarbon receptor (AhR) and is required for receptor function. The presence of AhR in Group B tumors underlines the role that chemicals play in the disease. In a single report concerning AhR, it was reported that genetic variations are linked to increased risk of BC [21].
Chromosomal-based analysis of transcription factors
Since gene expression is regulated in a chromosomal-dependent pattern, we separated the binding sites of TFs in order to find patterns of TFBMs among all five studied groups.
Our chromosomal-dependent analysis posed the following three questions: 1) Are chromosomal gene expression levels relative to the number of genes? 2) Is the number of the annotated TF binding sites relative to the number of DE genes? 3) Is the number of the annotated TF binding sites relative to the total chromosomal expression levels? Based on our analysis, the answer to these three questions appears to be “no”, at least for the majority of the chromosomes.
Chromosome 2
On chromosome 2, a linearity was observed between the number of DE genes and the number of TF binding sites with respect to the groups (R2 = 0.92). Yet, the order of the variables did not coincide with the grade of the tumor. In particular, local minima and maxima were represented by tumors of Groups A and B, respectively, while tumors of Group C (muscle-invasive T2-3, grade III) and metastatic tumors group were found in between. In some reports, it was shown that aberrations in chromosome 2 participate in alternative tumors of the bladder, such as lymphomas [22,23], with a particular role of CHOP (C/EBP homologous protein). Also, AP1 (activator protein 1) whose function with NFκB and positive regulation of tumor progression is linked to loss of SPAPC [24], was predicted by our analysis. Finally, MYC was uniquely annotated among metastatic TCCs, underlining its recent suggestions for therapeutic targeting in the disease [25,26]. Interestingly, chromosome 2 followed a linear pattern between “TFs” and “Genes”, irrespective of the tumor group, probably implying a wider role in the genesis and progression of the disease.
Chromosome 11
A similar behavior was observed in chromosome 11, but with a decreasing tendency (the more TF binding sites we annotated, the less the number of DE genes on the chromosome was found). This was again irrespective of the tumor group, where the local minima and maxima were represented by Groups C and A. Of note, AP1 was annotated in Group A, which also coincided with the functions’ local maxima as in the case of chromosome 2. At the same time, we predicted GATA1 and GATA3 among tumors of Group C, both related to poor prognosis and invasiveness in BC [27]. On the other hand, estrogen receptor (ER) was predicted to regulate metastatic tumors. Low ER expression was recently related to tumor aggressiveness [28]. Also, hormonal receptors are considered to be attractive therapeutic targets, although BC is not considered an endocrine-related neoplasia [29].
Chromosome 15
On chromosome 15, metastatic tumors did not have uniquely annotated TF binding sites, while ER and P53 were annotated in tumors of Group A. AR was uniquely annotated in Group C tumors, implying a hormonal-dependent function for this chromosome.
Chromosome 17
Chromosome 17 manifested a very interesting behavior. Regression analysis revealed that metastatic samples were separated from the rest three groups, while the three groups manifested a perfect linear correlation, with descending order, moving from more (Group C-muscle-invasive) to less aggressive tumor types (Group A/non-muscle invasive). The metastatic cancers had several uniquely annotated binding sites for the following TFs; NFκB, PAX5, YY1 and PPARA. High NFκB expression has been previously related to BC, but not to its stage [30]. Its expression has been linked to prognosis and response to therapy [8,31], as well as tumor progression [32]. PPARG has been previously reported to play significant role in tumor invasion [17]. Our analysis underlines the paramount importance of PPARs in the progression and aggressiveness of BC. It’s worth mentioning that NFκB was present in the two most aggressive tumor groups (Group C/muscle-invasive T3-grade III and metastatic tumors) and was absent in the other two groups (non-muscle invasive TCCs), suggesting an important role in tumor aggressiveness.
2021 Copyright OAT. All rights reserv
Chromosome 18
Chromosome 18 was also an interesting case, since the correlation between the number of DE genes and gene expression levels was marginally linear. This was actually the only chromosome that manifested a linear relation between the chromosomal mean expression levels and the number of DE genes. In this case, the unknown factor was the predicted TF binding sites, since such a behavior could be attributed to them. To our surprise, in the case of chromosome 18 there were a few unique TF binding sites identified in each group. In particular, non-muscle invasive tumors of Group A had annotated the homeobox protein NKX-2.5 (previously reported to be the unique discriminator of the neurokinin 1 receptor), accommodating the translocation of NFκB to the nucleus [33]. Furthermore, muscle-invasive TCCs of Group C had three uniquely annotated binding sites for the following TFs: GR (glucocorticoid receptor), CP2 and ZIC2. The GR is interesting, since it is questionable whether the co-administration of chemotherapy and glucocorticoids (GCs) should be recommended and whether GCs interfere with chemotherapy administration and subsequently with tumor survival. It was previously reported that the induction of GR promotes cell proliferation and resistance, but at the same time it inhibits cell invasion and probably metastasis. Yet, it appears that the activation of GR and its effects are GC-dependent as it has been shown that different GCs have different impact on cell survival and apoptosis [34]. The fact that the mean chromosomal expression is negative (compared to the controls) reinforces the finding of the GR in muscle-invasive TCCs of group C. The current findings suggested a special role for chromosome 18 in TCC of the urinary bladder and the predicted TFs could be used as putative therapeutic targets.
Chromosome X
A similar pattern to chromosome 17 was manifested in chromosome X. Chromosome X presented linear correlations between the number of “TFs”, the number of DE genes and the “TF/Genes” ratio. Yet, gene expression along with additional variables did not present any significant relations. However, when looking closer to the patterns of variables in X chromosome, we found that non-muscle invasive TCCs (groups A and B) and muscle-invasive TCCs (group C), present linear dynamics with respect to the mean chromosomal expression and the ratio binding sites TF/Genes. As in the case of chromosome 17, genes on chromosome X appear to be suppressed with regards to the average expression levels. Group A tumors had uniquely annotated binding sites of TFs such as HOXA9 and MEIS1, two proteins known for their role in acute leukemia but not BC. Yet, few reports mention a role for HOXA9 in tumor invasiveness and progression [35]. PAX6 was annotated in chromosome X for TCCs of Group A and it was reported to play an import role in tumor suppression [36]. Furthermore, CDC5 (cell division cycle 5) was uniquely annotated among Group C tumors. CDC5 is a cell cycle regulator, whose phosphorylation also controls RNA processing [37]. In a previous report we showed that CDC20 is consistently up-regulated in all the TCC samples that we examined, while both CDC20 and CDC5 appear to control mitotic phase in eukaryotic cells [8,38]. In the group of metastatic samples, NF1 (neurofibromin 1), which is involved in metastatic, invasive bladder cancer [39] was annotated. The nuclear factor, erythroid 2-like 2 (NRF2) was another uniquely annotated TF in chromosome X. NRF2 has been reported as a factor for chemotherapy resistance and whose expression is linked to the activation of antioxidant genes [40]. The paramount importance of chromosome X in the progression and ontogenesis of this disease has also been previously highlighted by others [41].
Concluding remarks
From the present work, it is evident that TFBM analysis through computational analyses can successfully and reliably predict known TFs in urinary bladder cancers of different histology. This approach could prove useful in the understanding of the disease, due to its ability to investigate massive amounts of data and extract summaries of information. Here, we found uniquely annotated TFs among non-muscle invasive and muscle-invasive TCCs of low and high grade. We highlight chromosomes 2, 11, 15, 17, 18 and X, reporting that they manifest linear dynamics with respect to computed variables including the mean chromosomal expression, the number of TF binding sites and the number of DE genes. In particular, since ER and PR were uniquely identified among different tumor groups, our computational analyses could predict a hormone-dependent mechanism underlying bladder cancer. In addition, it appears that the inflammatory machinery plays an important role in the progression of the disease, through NFκB and AP1. Finally, we highlight the importance of the control of the cell cycle in TCC through CDC5.
Conflict of interest statement
The authors state that they do not have any financial interests or connections, direct or indirect that might raise the question of bias in the present work.
Author contributions
AZ and GIL designed and performed the experimental procedures and data interpretation, prepared and submitted the manuscript. MB assisted in data interpretation and in drafting the manuscript.
References
- Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, et al. (2007) Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 18: 581-592. [Crossref]
- van Rhijn BW, Burger M, Lotan Y, Solsona E, Stief CG, et al. (2009) Recurrence and progression of disease in non-muscle-invasive bladder cancer: from epidemiology to treatment strategy. Eur Urol 56: 430-442. [Crossref]
- Sylvester RJ, van der Meijden AP, Oosterlinck W, Witjes JA, Bouffioux C, et al. (2006) Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: a combined analysis of 2596 patients from seven EORTC trials. Eur Urol 49: 475-477. [Crossref]
- Lopez-Beltran A, Montironi R (2004) Non-invasive urothelial neoplasms: according to the most recent WHO classification. Eur Urol 46: 170-176. [Crossref]
- Dyrskjøt L, Kruhøffer M, Thykjaer T, Marcussen N, Jensen JL, et al. (2004) Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res 64: 4040-4048. [Crossref]
- Cohen BA, Mitra RD, Hughes JD, Church GM (2000) A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet 26: 183-186. [Crossref]
- Lambrou GI, Adamaki M, Delakas D, Spandidos DA, Vlahopoulos S, et al. (2013) Gene expression is highly correlated on the chromosome level in urinary bladder cancer. Cell Cycle 12: 1544-1559. [Crossref]
- Zaravinos A, Lambrou GI, Boulalas I, Delakas D, Spandidos DA (2011) Identification of common differentially expressed genes in urinary bladder cancer. PLoS One 6: e18135. [Crossref]
- Zaravinos A, Lambrou GI, Volanis D, Delakas D, Spandidos DA (2011) Spotlight on differentially expressed genes in urinary bladder cancer. PLoS One 6: e18255. [Crossref]
- Altman NS, Hua J (2006) Extending the loop design for two-channel microarray experiments. Genet Res 88: 153-163. [Crossref]
- Mengual L, Burset M, Ars E, Lozano JJ, Villavicencio H, et al. (2009) DNA microarray expression profiling of bladder cancer allows identification of noninvasive diagnostic markers. J Urol 182: 741-748. [Crossref]
- Monzon FA, Lyons-Weiler M, Buturovic LJ, Rigl CT, Henner WD, et al. (2009) Multicenter validation of a 1,550-gene expression profile for identification of tumor tissue of origin. J Clin Oncol 27: 2503-2508. [Crossref]
- Cole SW, Yan W, Galic Z, Arevalo J, Zack JA (2005) Expression-based monitoring of transcription factor activity: the TELiS database. Bioinformatics 21: 803-810. [Crossref]
- Zhang B, Kirov S, Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33: W741-748. [Crossref]
- Zhang B, Schmoyer D, Kirov S, Snoddy J (2004) GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics 5: 16. [Crossref]
- Beisvag V, Jünge FK, Bergum H, Jølsum L, Lydersen S, et al. (2006) GeneTools--application for functional annotation and statistical hypothesis testing. BMC Bioinformatics 7: 470. [Crossref]
- DeGraff DJ, Cates JM, Mauney JR, Clark PE, Matusik RJ, et al. (2013) When urothelial differentiation pathways go wrong: implications for bladder cancer development and progression. Urol Oncol 31: 802-811. [Crossref]
- Wang Y, Tan H, Xu D, Ma A, Zhang L, et al. (2014) The combinatory effects of PPAR-γ agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int J Mol Med 34: 262-268. [Crossref]
- Lee HJ, Zhuang G, Cao Y, Du P Kim HJ, et al. (2014) Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26: 207-221. [Crossref]
- Zhang B, Lu Z, Hou Y, Hu J, Wang C (2014) The effects of STAT3 and Survivin silencing on the growth of human bladder carcinoma cells. Tumour Biol 35: 5401-5407. [Crossref]
- Figueroa JD, Malats N, García-Closas M, Real FX, Silverman D, et al. (2008) Bladder cancer risk and genetic variation in AKR1C3 and other metabolizing genes. Carcinogenesis 29: 1955-1962. [Crossref]
- Bosse KR, Shukla AR, Pawel B, Chikwava KR, Santi M, et al. (2014) Malignant rhabdoid tumor of the bladder and ganglioglioma in a 14 year-old male with a germline 22q11.2 deletion. Cancer Genet 207: 415-419. [Crossref]
- Partridge EA, Canning D, Long C, Peranteau WH, Hedrick HL, et al. (2014) Urologic and anorectal complications of sacrococcygeal teratomas: prenatal and postnatal predictors. J Pediatr Surg 49: 139-142. [Crossref]
- Said N, Frierson HF, Sanchez-Carbayo M, Brekken RA, Theodorescu D (2013) Loss of SPARC in bladder cancer enhances carcinogenesis and progression. J Clin Invest 123: 751-766. [Crossref]
- Jeong KC, Kim KT, Seo HH, Shin SP, Ahn KO, et al. (2014) Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. J Urol 191: 510-518. [Crossref]
- Lv L, Deng H, Li Y, Zhang C, Liu X, et al. (2014) The DNA methylation-regulated miR-193a-3p dictates the multi-chemoresistance of bladder cancer via repression of SRSF2/PLAU/HIC2 expression. Cell Death Dis 5: e1402. [Crossref]
- Liang Y, Heitzman J, Kamat AM, Dinney CP, Czerniak B, et al. (2014) Differential expression of GATA-3 in urothelial carcinoma variants. Hum Pathol 45: 1466-1472. [Crossref]
- Miyamoto H, Yao JL, Chaux A, Zheng Y, Hsu I, et al. (2012) Expression of androgen and oestrogen receptors and its prognostic significance in urothelial neoplasm of the urinary bladder. BJU Int 109: 1716-1726. [Crossref]
- Miyamoto H, Zheng Y, Izumi K (2012) Nuclear hormone receptor signals as new therapeutic targets for urothelial carcinoma. Curr Cancer Drug Targets 12: 14-22. [Crossref]
- Ozbek E, Otunctemur A, Calik G, Aliskan T, Cakir S, et al. (2011) Comparison of p38MAPK (mitogene activated protein kinase), p65 NFkappaB (nuclear factor kappa b) and EMMPRIN (extracellular matrix metalloproteinase inducer) expressions with tumor grade and stage of superficial and invasive bladder tumors. Arch Ital Urol Androl 83: 181-187. [Crossref]
- Koga F, Yoshida S, Tatokoro M, Kawakami S, Fujii Y, et al. (2011) ErbB2 and NFκB overexpression as predictors of chemoradiation resistance and putative targets to overcome resistance in muscle-invasive bladder cancer. PLoS One 6: e27616. [Crossref]
- Levidou G, Saetta AA, Korkolopoulou P, Papanastasiou P, Gioti K, et al. (2008) Clinical significance of nuclear factor (NF)-kappaB levels in urothelial carcinoma of the urinary bladder. Virchows Arch 452: 295-304. [Crossref]
- Saban, R., et al., (2007) Bladder inflammatory transcriptome in response to tachykinins: neurokinin 1 receptor-dependent genes and transcription regulatory elements. BMC Urol 7: 7. [Crossref]
- Ishiguro H, Kawahara T, Zheng Y, Kashiwagi E, Li Y, et al. (2014) Differential regulation of bladder cancer growth by various glucocorticoids: corticosterone and prednisone inhibit cell invasion without promoting cell proliferation or reducing cisplatin cytotoxicity. Cancer Chemother Pharmacol 74: 249-255. [Crossref]
- Kim YJ, Yoon HY, Kim JS, Kang HW, Min BD, et al. (2013) HOXA9, ISL1 and ALDH1A3 methylation patterns as prognostic markers for nonmuscle invasive bladder cancer: array-based DNA methylation and expression profiling. Int J Cancer 133: 1135-1142. [Crossref]
- Sacristan R, Gonzalez C, Fernández-Gómez JM, Fresno F, Escaf S, et al. (2014) Molecular classification of non-muscle-invasive bladder cancer (pTa low-grade, pT1 low-grade, and pT1 high-grade subgroups) using methylation of tumor-suppressor genes. J Mol Diagn 16: 564-572. [Crossref]
- Gräub R, Lancero H, Pedersen A, Chu M, Padmanabhan K, et al. (2008) Cell cycle-dependent phosphorylation of human CDC5 regulates RNA processing. Cell Cycle 7: 1795-1803. [Crossref]
- Rudner AD, Murray AW (2000) Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J Cell Biol 149: 1377-1390. [Crossref]
- Ross JS, Wang K, Al-Rohil RN, Nazeer T, Sheehan CE, et al. (2014) Advanced urothelial carcinoma: next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod Pathol 27: 271-280. [Crossref]
- Ye P, Mimura J, Okada T, Sato H, Liu T, et al. (2014) Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol 34: 3421-3434. [Crossref]
- Rahmani AH, Alzohairy M, Babiker AY, Khan AA, Aly SM, et al. (2013) Implication of androgen receptor in urinary bladder cancer: a critical mini review. Int J Mol Epidemiol Genet 4: 150-155. [Crossref]
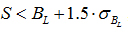 , where S is the measured signal intensity, BL is the local background measured and σBL is the standard deviation of the local background. Background correction was performed by subtracting the median global background from the median local background from the signal intensity. A threshold of 2 was set as cut-off, meaning that spot intensity should be twice as much as that of the background. Microarray data were cross-normalized, using a quantlie algorithm, in order to account for the bias that is included due to experimentation, different platforms and different sampling. In order to compare all the available microarray datasets among them, we used the following methodology:
, where S is the measured signal intensity, BL is the local background measured and σBL is the standard deviation of the local background. Background correction was performed by subtracting the median global background from the median local background from the signal intensity. A threshold of 2 was set as cut-off, meaning that spot intensity should be twice as much as that of the background. Microarray data were cross-normalized, using a quantlie algorithm, in order to account for the bias that is included due to experimentation, different platforms and different sampling. In order to compare all the available microarray datasets among them, we used the following methodology: