Abstract
Objective
Until today there is no isolated molecular or clinical test to demonstrate or rule outMarfan syndrome (MFS). Diagnosis relies on the revised Ghent Criteria and genetic analysis ofFBN1 mutation. In fact, althoughFBN1 mutation is the cause of classical MFS there are patientswhere it could not be detected. Therefore this study evaluates whether there is a difference concerning Marfan phenotype between pediatric patients with FBN1 mutation and those where mutation could not be analyzed. Finally we estimate necessary differentiation concerning follow-up, prophylaxis and therapy between patients with clinical and those with genetic diagnosis.
Methods
We investigated 244 children (10.8 ± 5.4 y) with confirmed or assumed MFS. Theywere subjected to a standardized diagnostic program including echocardiography, examination according to revised Ghent Criteria and genetic analysis.
Results
We established MFS as diagnosis in 96 patients. Genetic analysis was performed in 84patients with demonstration of a causative FBN1 mutation in 62 patients (74%). We did not find differences concerning prevalence or age of manifestation of dilatation of sinus of Valsalva, ectopialentis or systemic manifestation by comparing patients with or without FBN1 mutation. Especially comparison of different types of mutation did not show significant difference.
Conclusion
For optimal diagnosis we need as well as clinical diagnosis according to revisedGhent Criteria genetic analysis. Furthermore clinical manifestations did not show significant differences between patients with or without FBN1mutation. We conclude that in pediatric populations of MFS, demonstration of a causative FBN1 gene mutation may not relate to different phenotypes. There is no need to differentiate between pediatric patients with and without detected FBN1mutation concerning follow-up, prophylaxis and therapy.
Key words
FBN1; Genotype; Marfan syndrome; Phenotype; Revised Ghent Criteria.
Introduction
Marfan syndrome (MFS) is an autosomal dominantly inherited connective tissue disorder with a wide range of clinical manifestations. It is caused by heterozygousFBN1 (fibrillin-1) mutations [1]. Manifestations of the cardiovascular system are the most important and life-limiting symptoms which need early enough medical or surgical treatment [2]. Other symptoms involve skeletal system, ocular system, skin, lung and cerebral system [3]. Unfortunately until today there is no definite single molecular or clinical test to demonstrate or rule out MFS [4].
Clinical diagnosis relies on Ghent Criteria (Ghent-1) since 1996 and revised Ghent Criteria (Ghent-2) since 2010. Ghent-2 includes symptoms of MFS and diagnoses MFS in different constellation of manifestation [5, 6]. Because of a wide range and variable age of onset of symptoms clinical diagnosis with Ghent-2 is not always possible [7]. Especially in toddlers and children diagnosis is a challenge [8]. To improve diagnosis in childhood a Kid-Short Marfan-Score (Kid-SMS) based on clinical symptoms was developed which is used to do risk stratification for probability of MFS after first consultation [9].
The clinical diagnosis or suspicion of MFS can be confirmed by identification of a causativeFBN1 mutation [10]. Even though there are more than 1200FBN1mutations described up todate (Universal Marfan database, UMD-FBN1, http://www.umd.be), there are patients with a clinical diagnosis whereFBN1mutation cannot be found [11]. In fact only in 66 to 91% of patients with MFS a causative FBN1 mutation is detected [12, 13]. Suggesting that alterations in other genes involved in connective tissue disorders and/or thoracic aortic aneurysms and dissections (TAADs) are responsible for the Marfan phenotype in FBN1-mutation negative patients. Accordingly there are patients with typical symptoms of MFS and genetic analysis where mutation in transforming growth factor beta receptor 1 (TGFBR1) or TGFBR2 was found. Besides, there are patients who neither show FBN1 mutation nor mutation in TGFBR1 orTGFBR2.
Due to the fact that there are patients without causative FBN1 mutation this present study should evaluate the effect of causative FBN1 mutation on clinical manifestation in children with MFS. It then should demonstrate whether it is necessary to differentiate patients with FBN1 mutationand those without but clinical diagnosis according Ghent-2 concerning prophylaxis, follow-up and therapy.
Methods
Patient cohort
We investigated 244 probands (10.84 ± 5.39 years) with confirmed or assumed MFS in our Marfan Clinic. All patients were subjected to a standardized diagnostic program including clinical examination, examination according to the Ghent-2, echocardiography and whenever possible and useful MRI scans at the University Heart Center Hamburg [14]. Genetic analysis was done whenever MFS was suspected by those investigations. To assess clinical data and samples we obtained informed consent of patients or parents of patients. The study was approved by the Hamburg ethical board (Project identification code: PV 4005).
Genetic analysis
We isolated genomic DNA from blood samples by standard procedures. The 65 coding exons and flanking intronic sequences of theFBN1gene (GenBank accession no. NM_000138.4) were amplified from genomic DNA of the patients. Primer sequences are available on request. We directly sequenced amplicons using the ABI BigDye Terminator Sequencing Kit (Applied Biosystems, Weiterstadt, Germany) and an automated capillary sequencer (ABI 3500; Applied Biosystems, Weitersnosologytadt, Germany). Sequence electropherograms were analyzed using the Sequence Pilot software (JSI medical systems, Kippenheim, Germany). All FBN1nucleotide changes fulfilled the criteria for causal FBN1 mutations according to the revised Ghent criteria.[5]. We classifiedFBN1 mutations as follows: missense mutations, nonsense mutations, mutations affecting splice sites, frameshift mutations, andin-frame mutations (small deletions). We analyzed for possible gross deletions/duplications in FBN1by multiplex ligation-dependent probe amplification according to the manufacturer’s protocol (MLPA; SALSA® MLPA® kit, probemix P065 and P066, MRC Holland, Amsterdam, Netherlands).
Clinical examination
To estimate the effect of FBN1 mutation on clinical manifestation of patients with Marfan syndrome we analyzed prevalence and age of manifestation of the three clinical cardinal symptoms of Ghent-2. We performed echocardiography with General Electric Vivid 7 with 10, 5 and 3 MHz probes to assess aortic root dilatation. We therefor operated in parasternal long axis view on 2D and M-Mode images using leading edge to leading edge technique at end diastole. We evaluated dilatation of sinus of Valsalva according to Roman et al.[15]. After all we compared prevalence and age of manifestation between patients with causative FBN1mutation and those without. In addition we analyzed type of mutation to evaluate whether there are differences between those.
Statistical analysis
We collected data using Filemaker software V.10 pro advanced. A subsequent statistical analysis was done with SPSS V16.0. Tables and figures were established with Microsoft Excel 2003 and SPSS V16.0. Quantitative variables were presented as means with standard deviation and prevalence in percentage. Means between groups were compared using unpaired t test, whereas all p values were two sided. P values <0.05 were considered to be statistically significant. Clinical differences were tested using log-rank test. Again p values <0.05 were considered significant.
Results
Until today 96 pediatric patients were diagnosed with MFS (96/244, 39%). 49 patients were clinically diagnosed according to Ghent-2 and had a causative FBN1mutation. A pathogenicFBN1mutation was found in 13 patients without a clinical diagnosis of MFS. Only clinicaldiagnosis with Ghent-2 was assessed in 34 patients, whereas in 22 patients FBN1analysis was negative and in 12 patients genetic analysis was not performed.
In fact, we performed genetic analysis in 84 of 244 patients. In total, 62 patients (62/84, 73.8%) had a causative FBN1 mutation. Genetic analysis in the other 22 (26.2%) patients who were diagnosed according to Ghent-2 did not reveal a FBN1mutation (Figure. 1). In detail genetic analysis showed 29 missense mutations, 11 splicing mutations, 9 frameshift mutations, 4 nonsense mutations and 3 in-frame mutations (small deletion) (Figure. 2).In the mutation-positive patients, dilatation of sinus of Valsalva was found in 67.9% (57/84) with a mean age of 9.5 ± 5.3 years, ectopialentis in 21.4% (18/84) with a mean age of 6.7 ± 4.5 years and systemic manifestation in 50.0 % (42/84) with a mean age of 12.3 ± 4.7 years.
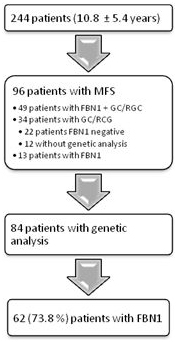
Figure 1. Flow chart of diagnosis.
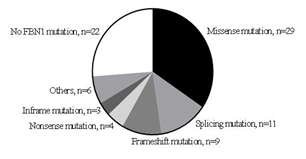
Figure 2. Mutation analysis in patients with Marfan syndrome.
Comparing patients with FBN1 mutation and those without concerning prevalence and age of manifestation of aortic root dilatation, ectopialentis and systemic manifestation there was no significant difference (Table 1).
|
|
Patients with FBN1
|
Patients without
|
Patients with FBN1
|
Patients without
|
|
|
|
|
mutation
|
FBN1 mutation
|
mutation
|
FBN1 mutation
|
p-
|
|
|
|
|
|
|
|
|
|
|
|
value
|
|
|
|
Prevalence (%)
|
Prevalence (%)
|
Age (years ± SD)
|
Age (years ± SD)
|
|
|
|
|
|
|
|
|
|
|
|
SV
|
66.1% (41/62)
|
68.2% (15/22)
|
8.9 ± 0.8
|
11.1 ± 1.5
|
0.1711
|
|
|
|
|
|
|
|
|
|
|
EL
|
24.2% (15/62)
|
13.6% (3/22)
|
7.2 ± 1.2
|
3.9 ± 0.2
|
0.2588
|
|
|
|
|
|
|
|
|
|
|
SysM
|
40.3% (25/62)
|
77.3% (17/22)
|
11.3 ± 1.0
|
13.7 ± 0.9
|
0.0924
|
|
|
|
|
|
|
|
|
|
Table 1. Prevalence and age of manifestation of dilatation of sinus of Valsalva (SV), ectopialentis (EL) and systemic manifestation (SysM) in patients with and without FBN1 mutation.
We also analyzed mutations in detail and compared patients with missense (n=29), frameshift (n=9) and nonsense (n=4) and splicing mutations (n=11) concerning prevalence and age of manifestation of clinical symptoms. Similarly, we did not find a significant difference between the groups of mutations (Table 2 and 3). Patients with frameshift or nonsense mutation did not show ectopialentis in this present study group.
|
|
Patients with
|
Frameshift or
|
Patients
|
with
|
Splicing
|
Patients
|
with
|
Missense
|
|
|
|
Nonsense
|
mutation,
|
mutation,
|
|
|
mutation,
|
|
|
|
|
|
Prevalence (%)
|
Prevalence (%)
|
|
Prevalence (%)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SV
|
84.6% (11/13)
|
|
81.8% (9/11)
|
|
62.1% (18/29)
|
|
|
|
|
|
|
|
|
|
|
|
|
EL
|
0 (0/13)
|
|
36.4% (4/11)
|
|
34.5% (10/29)
|
|
|
|
|
|
|
|
|
|
|
|
|
SysM
|
61.5% (8/13)
|
|
27.3% (3/11)
|
|
37.9% (11/29)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table 2. Prevalence of dilatation of sinus of Valsalva (SV), ectopia lentis (EL) and systemic manifestation (SysM) in patients with frameshift or nonsense, splicing and missense mutation.
|
|
Patients with Frameshift or
|
Patients
|
with
|
Splicing
|
Patients with Missense
|
|
|
Nonsense mutation
|
mutation,
|
|
|
mutation
|
|
|
Age (years ± SD)
|
Age (years ± SD)
|
|
Age (years ± SD)
|
|
|
|
|
|
|
|
|
SV
|
9.38 ± 1.58
|
9.13 ± 2.08
|
|
|
7.68 ± 1.04
|
|
|
|
|
|
|
|
|
EL
|
-
|
10.12 ± 3,20
|
|
|
6.05 ± 1.29
|
|
|
|
|
|
|
|
|
SysM
|
12.27 ± 1.56
|
11.01 ± 3.54
|
2021 Copyright OAT. All rights reserv
|
|
10.39 ± 1.71
|
|
|
|
|
|
|
|
Table 3. Age of manifestation of dilatation of sinus of Valsalva (SV), ectopia lentis (EL) and systemic manifestation (SysM) in patients with frameshift or nonsense, splicing and missense mutation.
Discussion
The care of patients with MFS is a challenge for all involved disciplines. Due to the fact that there are pediatric patients with MFS with causativeFBN1 mutation and some without, the question comes up whether it is necessary to distinguish those two patient groups in a special way concerning follow-up, prophylaxis and therapy [16].
First, this study showed a causative FBN1mutation in 74% of patients which is similar to the detection rate of FBN1 mutation described in previous articles [12,13]. There are certainly still unknown causative FBN1 mutations or other genes with loss of function inducing MFS [12].TGFB2, TGFBR1andTGFBR2are genes which cause very similar clinical symptoms to MFSand induce Loeys-Dietz-Syndrome (LDS) and Marfan syndrome type 2 (MFS2). In the majority of FBN1-negative patients in this study mutation testing of TGFB2, TGFBR1 and TGFBR2was also done and did not show an identification of mutation. In patients where genetic analysis was done before 2005 where LDS caused by mutation ofTGFBR1 or TGFBR2 was published we did a re-assessment of mutation analysis subsequently [17]. Other gene mutations than FBN1,TGFB2, TGFBR1andTGFBR2were not analyzed routinely [11,18]. In a small part of resultstechnical limitations and variation of screening methods in different centers may be the reason for not detecting FBN1mutation.
On the other side we examined children who only showed causative FBN1 mutation without diagnosis according to Ghent-2 which was also documented by Faivreet al.[19]. FBN1 testing is important for diagnosis of Marfan syndrome as there are children without enough clinical symptoms or a mild form of MFS [16]. Especially in children with an age-dependent onset of symptoms genetic analysis is indispensible. Moreover there is a real benefit of molecular analysis in children with pathological mutation known in the family. It thus allows to screen relatives of Marfan patients before the occurrence of symptoms or even prenatal or directly postnatal. It is then possible to decide whether monitoring of the child has to be continued or can be stopped because of no inherited mutation [20].
Thus again we showed that there are two very important ways to diagnose MFS which both cover different patient groups. Neither clinical diagnosis nor genetic analysis alone shows the sensitivity that the combination of both does. In the future it may be useful to repeat genetic analysis in FBN1-negative patients to evaluate new gene mutations or benefit from technical advances but currently clinical diagnosis is not dispensable.
Secondary, to estimate the manifestation of symptoms in pediatric patients with clinical Marfan diagnosis and those with causative FBN1mutation and to decide whether there is a need to distinguish this patient groups, we analyzed the three cardinal symptoms of Ghent-2. Thereby dilatation of sinus of Valsalva was the most common symptom similar to other studies [21-23] and ectopialentis was the earliest symptom as shown before [22].
We did not identify a significant difference concerning prevalence or age of manifestation aortic root dilatation, ectopialentis and systemic manifestation in patients with and those without causative FBN1mutation but Marfan phenotype according to Ghent-2. We also did not show a significant difference by comparing different mutation types. As soon patients were diagnosed with MFS the risk of developing clinical symptoms, especially cardiac pathologies was equal in both groups independently from the way of diagnosis.
Accordingly, early diagnosis of MFS, self-contained whether clinical or genetic diagnosis is essential for further treatment and subsequently prevention of high mortality of untreated aortic complications in MFS. Particularly because the effectiveness of early pharmacological prophylaxis in pediaetric patients with MFS is established [24]. For optimal supply of Marfanpatients comprehensive clinical and genetic analysis is essential [25].
We conclude first, to guarantee best quality of diagnosis clinical diagnosis according to Ghent-2 and molecular analysis have to be combined as there are patients with Marfan genotype without clinical manifestation at the time of presentation and patients with clinical symptoms without causative FBN1 mutation.
We conclude secondarily, in clinical life there is no need to distinguish between pediatric patients with clinical diagnosis according to Ghent-2 or molecular diagnosis concerning prophylaxis, follow-up and therapy. As soon Marfan syndrome is diagnosed with a FBN1mutation or clinical diagnosis according to Ghent-2 every patient needs regular follow-up and individualized prophylaxis and therapy according to clinical symptoms and especially cardiac pathologies.
Study Limitations
This study has several limitations. The real age of onset of symptoms in first presentation to Marfan Clinic is very difficult. As soon as a patient presents to Marfan Clinic we suppose this point as the onset age of existing symptoms although in many cases the symptom probably existed before. Due to the long period of this retrospective study different tools were used to analyzeFBN1 mutation. Besides, after splitting patients with different mutations into different groups for comparison groups were pretty small. To confirm the results of this study a bigger group of patients should be analyzed. Finally it is well known that there are differences between detailed location of mutation, e.g. exon 24 to 32 [21]. We did not analyze this because of a too small group of patients.
Authorship and contributorship
VCS conceptualised the study, performed data collection, designed data collection instruments and performed initial analysis, drafted the initial manuscript and approved final manuscript. GCM supervised data collection, performed initial analysis and critically reviewed and revised the manuscript and approved the final manuscript. KJS and GH coordinated and performed data collection, designed data collection instruments, critically reviewed the manuscript and approved the final manuscript. FA and MR performed data collection, critically reviewed the manuscript and approved the final manuscript. KK carried out molecular genetic analysis, critically reviewed the manuscript and approved the final manuscript. BK carried out molecular genetic analysis andapproved the final manuscript. YvK and RK reviewed and revised the manuscript andapproved the final manuscript. TSM conceptualised the study, coordinated and supervised data collection, reviewed, revised and approved the final manuscript.
Acknowledgements
We would like to thank Mrs Claudia Schlesner for documentation of data.
Competing interests
The authors declare that they have no competing interests.
References
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, et al. (1991) Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352: 337-339. [Crossref]
- Ammash NM, Sundt TM, Connolly HM (2008) Marfan syndrome-diagnosis and management. Curr Probl Cardiol 33: 7-39. [Crossref]
- Dean JC1 (2007) Marfan syndrome: clinical diagnosis and management. Eur J Hum Genet 15: 724-733. [Crossref]
- von Kodolitsch Y, Rybczynski M, Detter C, Robinson PN (2008) Diagnosis and management of Marfan syndrome. Future Cardiol 4: 85-96. [Crossref]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, et al. (2010) The revised Ghent nosology for the Marfan syndrome. J Med Genet 47: 476-485. [Crossref]
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE (1996) Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 62: 417-426. [Crossref]
- Faivre L, Collod-Beroud G, Adès L, Arbustini E, Child A, et al. (2012) The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet 81: 433-442. [Crossref]
- Lipscomb KJ, Clayton-Smith J, Harris R (1997) Evolving phenotype of Marfan's syndrome. Arch Dis Child 76: 41-46. [Crossref]
- Mueller GC, Stark V, Steiner K, Weil J, von Kodolitsch Y, et al. (2013) The Kid-Short Marfan Score (Kid-SMS) - an easy executable risk score for suspected paediatric patients with Marfan syndrome. Acta Paediatr 102: e84-89. [Crossref]
- Maslen CL, Glanville RW (1993) The molecular basis of Marfan syndrome. DNA Cell Biol 12: 561-572. [Crossref]
- Pees C, Michel-Behnke I, Hagl M, Laccone F (2014) Detection of 15 novel mutations in 52 children from 40 families with the Marfan or Loeys-Dietz syndrome and phenotype-genotype correlations. Clin Genet 86: 552-557. [Crossref]
- Loeys B, De Backer J, van Acker P, Wettinck K, Pals G, Nuytinck L, et al. (2004) Comprehensive molecular screening of FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat: 24: 140-146.
- Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A (2001) Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med 161: 2447-2454. [Crossref]
- Sheikhzadeh S, Sondermann C, Rybczynski M, Habermann CR, Brockstaedt L, et al. (2014) Comprehensive analysis of dural ectasia in 150 patients with a causative FBN1 mutation. Clin Genet 86: 238-245. [Crossref]
- Roman MJ, Devereux RB, Kramer-Fox R, O'Loughlin J (1989) Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol 64: 507-512. [Crossref]
- Sheikhzadeh S, Kade C, Keyser B, Stuhrmann M, Arslan-Kirchner M, et al. (2012) Analysis of phenotype and genotype information for the diagnosis of Marfan syndrome. Clin Genet 82: 240-247. [Crossref]
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, et al. (2006) Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 355: 788-798. [Crossref]
- Rommel K, Karck M, Haverich A, von Kodolitsch Y, Rybczinski M, Müller G, et al. (2005) Identification of 29 Novel and nine recurrent fibrillin-1 (FBN1) mutations and genotype-phenotype correlation in 76 patients with Marfan syndrome. Hum Mutat: 26: 529-539.
- Faivre L, Collod-Beroud G, Callewaert B, Child A, Binquet C, et al. (2009) Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24-32 mutation. Eur J Hum Genet 17: 491-501. [Crossref]
- Keane MG, Pyeritz RE (2008) Medical management of Marfan syndrome. Circulation 117: 2802-2813. [Crossref]
- Faivre L, Masurel-Paulet A, Collod-Beroud G, Callewaert BL, Child AH, Stheneur C, et al. (2009) Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics: 123: 391–398.
- Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, et al. (2007) Effect of mutation type and location on clinical outcome in ,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 81: 454-466. [Crossref]
- Mueller GC, Stark V, Steiner K, von Kodolitsch Y, Rybczynski M, et al. (2013) Impact of age and gender on cardiac pathology in children and adolescents with Marfan syndrome. Pediatr Cardiol 34: 991-998. [Crossref]
- Mueller GC, Stierle L, Stark V, Steiner K, von Kodolitsch Y, et al. (2014) Retrospective analysis of the effect of angiotensin II receptor blocker versus β-blocker on aortic root growth in paediatric patients with Marfan syndrome. Heart 100: 214-218. [Crossref]
- von Kodolitsch Y, Rybczynski M, Bernhardt A, Mir TS, Treede H, et al. (2010) Marfan syndrome and the evolving spectrum of heritable thoracic aortic disease: do we need genetics for clinical decisions? Vasa 39: 17-32. [Crossref]