Abstract
Lipids composition of cellular membrane is known to be crucial for regulations of activities of many cells. In immune cells, activities of diverse receptors including T cell receptors and innate immunity receptors are modulated by lipids. In the presence of cholesterol in membranes, saturated fatty acids (FAs) promote formation of ordered lipid microdomain formation and enhance receptor-mediated signaling and inflammation, but the mechanistic detail for the effects of saturated FAs has not been well understood. Here we show that, for a poly-Ile transmembrane (TM) helical peptide, a self-dimerized state is stabilized by about -2.2 kJ/mol by the lipid bilayer of 1:1:1 palmitoyloleoylphosphatidylcholine (POPC)/dipalmitoylphosphatidylcholine (DPPC)/cholesterol compared to a pure dioleoylphosphatidylcholine (DOPC) bilayer in molecular simulations with a united-atom force field. Energy decomposition analysis suggested that the DOPC bilayer has a high Lennard-Jones (LJ) potential energy cost due to non-optimal protein-lipid interactions (poor fitting) upon peptide dimerization relative to the monomers state, but the 1:1:1 bilayer showed a relatively small differential of the protein-lipid LJ potential between the dimer and monomer states. In the 1:1:1 bilayer, both POPC and DPPC resided near the dimerized peptides, with cholesterol mostly residing >3 Å distant from peptides surface. However, a potential energy decomposition analysis did not support the view that peptides segregation from the cholesterol-rich sub-compartment is important for stabilizing the dimerization. Rather, peptide solvation by phospholipids preserved relatively well even after dimerization possibly due to the straightened acyl chains was the key for the dimer stabilization. Taken together, saturated FAs of phospholipids and cholesterol appear to stabilize transmembrane helical peptide dimerization in a sequence-nonspecific manner through the ability of such lipids to indirectly assist peptides solvation by lipids after peptide dimerization.
Key words
protein clustering, protein recruitment, dynamics simulations, GROMOS, lipid rafts
Introduction
Dietary fatty acids (FAs) are incorporated into phospholipids of cell membranes and influence cellular functions [1,2]. Altered FA compositions (of phospholipids) of immune cells have been shown to modulate activities of the cells [1,3,4]. Polyunsaturated FAs (PUFAs) including n-3 PUFAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have been shown to inhibit T cell proliferation [5,6], whereas diets rich in saturated FAs have been implicated in the development of metabolic diseases, insulin resistance and atherosclerosis, in which low-grade inflammation is considered to be involved [7]. Ample data pointed to a view that saturated FAs and trans-FAs favor a pro-inflammatory state contributing to chronic inflammation, autoimmunity, allergy, atherosclerosis as well as other metabolic diseases [7]. In contrast, anti-inflammatory effects and overall benefits of n-3 PUFAs in such chronic inflammation have been well documented [7].
Mechanisms for differential effects of FAs have been under intensive researches. To name but a few, saturated FAs have been shown to induce cytokine secretion in T-cells in the absence of T-cell activation in a dose-dependent manner, while unsaturated FAs were unable to do so [8]. Shaikh et al. reported that saturated FAs-enriched diets increased T cell receptor nanoscale clustering in murine CD4+ T cells to a level usually seen on activated cells, whereas diets enriched in monounsaturated FAs or n-3 PUFA showed no such effect [9]. In Holzer et al. [10], consumption of high-fat diet caused the partitioning and activation of c-Src (a myristoylated protein) within detergent-insoluble subdomains of murine adipocytes and their in vitro analysis supported the results. Of note c-Src activation is a key event to c-Jun N-terminal kinase (JNK) activation, and the latter can be induced by saturated FA but not by PUFA [11]. We suggest review articles on the effects of FAs on immune cells [12-14].
Immunomodulatory effects of FAs are considered to be mediated by several modalities including: 1) interference with lipid mediator molecules, which involves cyclooxygenase as a key player, 2) specific receptor protein-mediated signaling leading to gene expression regulation, and 3) effects on physical properties of membrane, including formation of lipid-mediated microdomains in biological membranes [15,16]. For 2), various machineries ranging from Toll-like receptors (TLRs) to transcriptional machineries act in concert. Among this category are recent discoveries of G protein-coupled receptors (GPCRs) for n-3 FAs, such as GPR120 [17,18]. n-3 PUFAs (DHA and EPA), but not saturated FAs, exert potent anti-inflammatory effects through GPR120 [18]. The mode 3) postulates the effects through modification of the physicochemical properties of lipid membranes. Saturated FAs and cholesterol generally increase lipid order and rigidity, and this mode may be associated with heterogeneity of lipid membranes due to formation of microdomains. Upon cell stimulation, cholesterol- and sphingolipid-rich microdomains of the plasma membrane called lipid rafts merge into larger platforms, facilitating interactions of some receptors with their signaling molecules [19-21]. PUFA-treatment of Jurkat T cells led to increased PUFAs in phospholipids in not only in bulk membranes but also isolated lipid rafts (based on sucrose gradient fractionations) and led to displacement from lipid rafts of proteins [22]. It is well known that, cytoplasmic and transmembrane (TM) proteins that are enriched in lipid rafts are generally acylated with saturated FAs. Interestingly, in Stulnig et al. [22], an analysis on Lck and the linker for activation of T cells (LAT), two palmitoylated proteins suggested that the displacement of proteins from lipid rafts occurs in PUFA-treated T cells even when proteins are acylated with palmitoyl residues.
Dimerization is the common mechanism responsible for activation of most, if not all, single-spanning TM protein receptors [23]. In many cases, dimerization occurs after ligand binding and is followed by activation of protein kinase activity. Dimerization/oligomerization of multi-spanning receptor proteins including GPCRs has also been recognized as part of normal signaling process [24]. For example, luteinizing hormone (LH) receptor localizes to membrane-type lipid microdomains upon ligand binding [25]. In some cases, oligomerization of receptors is associated with their translocation to particular membrane microdomains. For example, both single-particle tracing and biochemical approaches strongly suggested that LH receptors unbound to ligands are located in non-rafts and, after ligand binding, translocate into lipid rafts. Thus, dimerization and recruitment of TLR4 into lipid rafts are likely to be two critical events, possibly coupled to each other, that lead to TLR4 activation. Oligomerization status may be an important factor controlling partition of receptor proteins into lipid rafts [21].
Regarding the effects of FAs on membrane protein interactions, however, it is not clear as yet to what extent indirect effects of FAs through modulation of lipid rafts properties and of inter-microdomain heterogeneity are important. As the cellular difference between the liquid-ordered (Lo) and the liquid disordered (Ld) phases is much smaller than in the synthetic systems [16,26], physicochemical effects of FAs on protein dimerization/multimerization might not entirely depend on the preformed differences between inside and outside of microdomains. That is, the pro-inflammatory effect of saturated FAs may be operating for membranes with a substantial range of lipid ordering and stiffness. However, it is generally difficult to address these questions using live cells, as the significance of recruitment to rafts as well as that of acylation by saturated SA differs among proteins [25].
We have recently been focusing on molecular dynamics (MD) simulation analysis of the free energy of TM dimerization using united-atom (UA) or all-atom (AA) force fields (FFs) [27,28]. A main challenge is requirement of intensive computation (see the Methods section), but, the use of united atom (UA) FFs can reduce the computation cost to ~20-25% of that with AA FFs. Here we carried out a free energy analysis of self-dimerization of a poly-Ile TM peptide under a UA FF. Marked stabilization of the dimerized state was observed in a saturated FA- and cholesterol-rich bilayer, compared with a pure DOPC bilayer. Although cholesterol was mostly resided distant from peptides and the peptides were solvated equally by both DPPC and POPC, it was not that strong lipid-lipid interactions characteristic of raft-like (saturated FA- and cholesterol-rich) bilayers facilitated the peptides segregation and dimerization. In fact, when the total potential energy was decomposed into the potential energy of peptide-peptide interaction, that of peptide-lipid interaction, and that of lipid-lipid interaction, the profile of lipid-lipid interaction potential energy did not support the scenario of the aforementioned segregation being the key player in the dimer stabilization. Rather, the potential energy term of lipid-protein interaction showed an intriguing feature; while in the DOPC bilayer the lipid-protein LJ term caused a high-energy cost upon peptide dimerization, in the raft-like bilayer it caused a relatively smaller energy cost (better solvation) upon peptide dimerization. Thus, the key factor for the dimer stabilization appeared to be some structural features, possibly involving high order parameters of lipid acyl chains characteristic to raft-like bilayers that enabled preserved solvation of peptides by lipids after dimerization and lowered the energy cost for dissolving peptides form lipids upon dimerization.
Methods - computational details
All molecular dynamics simulations were performed with the Gromacs suite version 4.5.4 [29]. The GROMOS53A6 was downloaded from the automated force field topology builder (ATB) site [30] and used as implemented in Gromacs for DOPC, POPC, DPPC, and cholesterol. Simple-point charge (SPC) water was used as provided by Gromacs. The initial structure and GROMOS53A6 parameters topology files for poly-Ile (i.e., (I)21) were prepared by modification of the files used in our recent analyses [27,28]. The N- and C-terminal structures used were -NH2 and -COOH, respectively. The temperature was kept constant at 323 K with Berendsen coupling and the semi-isotropic pressure coupling using Berendsen algorithm with 1 bar was used as in our recent paper [28]. Other conditions including the bond lengths constraint and the treatment of long-range Coulomb and LJ interactions were also the same as in [28].
The umbrella sampling simulation (based on the pull-code module of Gromacs) and subsequent merging via the weighted histogram analysis method (WHAM) [31] were used for the computation of the potential of mean force (PMF) profile for TM peptide dimerization using the method as previously described [27,28]. Briefly, for the umbrella sampling, a harmonic potential with a force constant of 3000 kJ/mol/nm2 on the distance (r) between the centers of mass of the helical peptide was applied. Sixteen initial windows with a spacing of 0.1 nm corresponding to the range of interhelical distances from 1.1 to 1.6 nm were generated. Then, two peptides were inserted in an antiparallel orientation into the equilibrated bilayers and overlapping lipids were manually moved. To reduce the influence of initial structures, eight independent structures were prepared for each interhelical distance r, for each set listed in Table 1. For the DOPC systems (#1), a 100-ns equilibration run was followed by a 400-ns production run. For the system of 1:1:1 POPC/DPPC/cholesterol (#2, Table 1), which we simply refer to as ‘the 1:1:1 bilayer’, a 100-ns equilibration run was performed prior to the production runs of 500 ns per window (Table 1). After using the WHAM method to compute the PMF profile GPMF(r), the dimerization free energy ∆Gdim for helical peptides was calculated as ∆Gdim=-RT lnKa , where Ka is the ratio of the time length during which the two peptides are in dimer to that during which they are in monomers. Ka is equivalent to the association constant estimated as Ka=[∫ πrg(r) dr]/Pm , where integration runs from 0 to Rc, with Rc being the cutoff that defines the dimerized state. g(r) is the two-dimensional radial distribution function (rdf) profile obtained by compensating the Boltzmann factor exp(-βGPMF(r)) with respect to the r-dependent increase in available phase space, and the normalization factor Pm is given as Pm=[ v/{π(Rmax2 - Rc2)}] * [∫ πrg(r) dr], where v is the bilayer area available to a peptide monomer at the standard concentration and the integration runs from Rc to Rmax. Of note, Pm represents the normalized v, that is, v weighted by the time length during which the two peptides are in monomers estimated by the integration. In the current study, Rc=1.6 nm and v=1.66 nm2 were used. On 30 Intel four-core 2.8 GHz central processing units (CPUs), a DOPC set of PMF analysis (500 ns/window, n=6) (do-S of Table 1) took ~60 days, while the computation time doubled for the 1:1:1 bilayer analysis. In the cases of occasional run-time errors due to severe collisions between molecules that halted simulations, the run was resumed using the coordinates of the 10 ns earlier time-frame and reseeding the velocities.
ID |
umbrella set name |
constituents |
simulation time |
∆Gdim (kJ/mol) |
1 |
Gr-Ile21-dopc /w |
do-S: 56 DOPC/2047 water |
6 × 500ns × 6 |
-0.01 |
2 |
Gr-Ile21-1-1-1/w |
1-1-1-raft: 24 POPC/24 DPPC 24 Chol/1835 water |
8 × 500ns × 6 |
-2.23 |
Table 1. Simulations systems and results
Results
Raft-like bilayer stabilizes dimerized state of poly-Ile TM helical peptides
To analyze the effect of lipid composition on peptide dimerization propensity, we performed UA simulations totaling 42 micro-seconds of the two identical poly-Ile peptides placed, in bilayers, at varied distances (separations) using umbrella-sampling methods (Figure 1) and obtained the profile of self-dimerization potential of mean force (PMF) (Figure 2). Our choice of the sequence (i.e., poly-Ile) was based on our recent findings that poly-Ile, but not Leu and Val, showed similar dimerization free energy profiles between GROMOS53A6 and CHARMM36 FFs (Nishizawa, manuscript preparation). As lipid membranes, we chose a DOPC bilayer and a bilayer of 1:1:1 POPC/DPPC/cholesterol that we refer to as the 1:1:1 bilayer (Table 1). The latter composition was chosen based on its similarity to the Niemela et al. [32] that used 1:1:1 POPC/palmitoyl-sphingomyelin (SM)/cholesterol (the system SA of Niemela et al. [32]) as well as on the corresponding experimental data showing miscibility (i.e., no phase boundary occurrence) down to 283 K [33]. Of note, based on the order parameter of C5-C7, the DOPC bilayer showed SCD=-0.20 and the 1:1:1 bilayer (as of POPC and DPPC) -0.29. The latter was found unchanged by the presence of the peptides. These show a degree of order lower than that of phospholipids of the system SA of Niemela et al. [32] that showed SCD of -0.41, but was higher than their 62:1:1 POPC/palmitoyl-SM/cholesterol (SCD=-0.18). So, our 1:1:1 bilayer was considered to have intermediate characteristics between DOPC bilayer and their system SA bilayer. We did not use SM based on our findings on the subtlety in headgroup and peptide interaction under UA FFs (Nishizawa, unpublished result) and on the limited information on SM
headgroup in this regard.

Figure 1. Representative snapshots of the self-dimerization PMF simulations of the poly-Ile peptide. (a) The DOPC system (#1 of Table 1). In this example the interhelical distance r was held at 1.3 nm. Representation scheme: cyan licorice, lipid acyl chains; spheres of middle size (red, and blue), phospholipid headgroup atoms (oxygen, and nitrogen atoms, respectively); small spheres (silver and red), water atoms; yellow bars, peptide backbone trace; green and cyan spheres, Ile side chains. Only lipid molecules located within a 2.5-nm-thick slice are shown. (b) The 1:1:1 bilayer system (#2 of Table 1) with r=1.6 nm. Represented as in (a), but cholesterol is shown in pink.
The obtained free energy profiles are shown (Figure 2). In the DOPC bilayer, the poly-Ile exhibited no propensity for self-dimerization as shown by a rather high free energy level at the distance between the peptides r=~1.3 nm relative to at r=1.6 nm. This corresponded to the dimerization free energy of -0.01 kJ/mol. Of note, compensation of the r-dependent increase in phase space (see Methods) led to such a non-positive value, which was somewhat counterintuitive. In contrast, the same peptides placed in the 1:1:1 bilayer showed the free energy profile with a well at r=1.2-1.3 nm. Integration yielded a dimerization energy of -2.23 kJ/mol. Standard errors for both curves (based on the six and eight trajectories for the DOPC and 1:1:1 bilayer, respectively, per umbrella window) were adequately small, and the binding free energy was significantly greater with the 1:1:1 bilayer than with the DOPC bilayer (t-test, P<0.01).
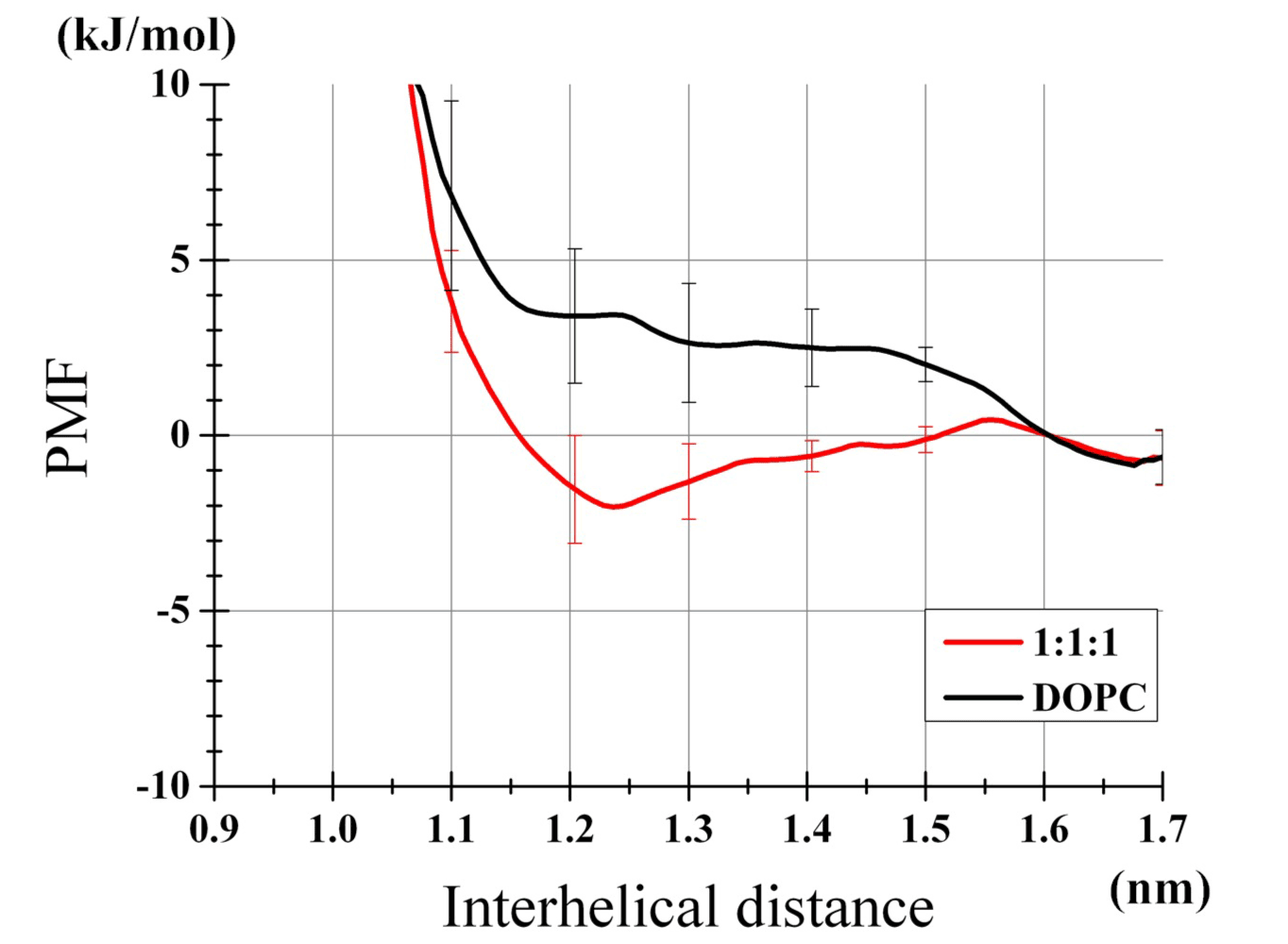
Figure 2. Profiles of TM helices dimerization PMF. Results of the DOPC bilayer and the 1:1:1 bilayer are shown. Error bars represent SEs from six (DOPC) and eight (1:1:1) independent umbrella analysis sets. The values relative to the values at r=1.6 nm are plotted.
Peptides were in contact with both POPC and DPPC but not with cholesterol in the raft-like bilayer
To examine distributions of lipids in the simulations, the radial distribution functions (rdfs) of lipids around peptides were computed for; 1) the DOPC bilayer with the interhelical distance r=1.3 nm (Figures 3A, 3B), 2) the DOPC bilayer with r=1.6 nm (Figures 3C, 3D), 3) the 1:1:1 bilayer with r=1.3 nm (Figures 3A, 3B), and 4) the 1:1:1 bilayer with r=1.6 nm (Figures 3C, 3D). Importantly, cholesterol in the 1:1:1 bilayer had very few direct contacts with peptides (<2.2 Å from peptides surface) in both dimerized and monomers states (Figures 3B, 3D). Another feature was that the distribution of DOPC in the proximity of peptides was largely comparable with the distribution of phospholipids (i.e., POPC and DPPC) in the 1:1:1 bilayer (Figures 3B, 3D). As a consequence of this, while the lipid-peptide interaction potential energy showed differences between the DOPC and the 1:1:1 systems (as shown below), it seemed impractical to explain this difference based on some metrics such as contacts or distributions. It was also noted that in our 1:1:1 system POPC showed higher density in the proximity of peptides than DPPC when peptides were dimerized (Figure 3B, black and gray lines), but the opposite trend was seen at r=1.6 nm (Figure 3D), supporting the view that the dimerization is associated with the peptides segregation from the cholesterol/DPPC-rich sub-areas.
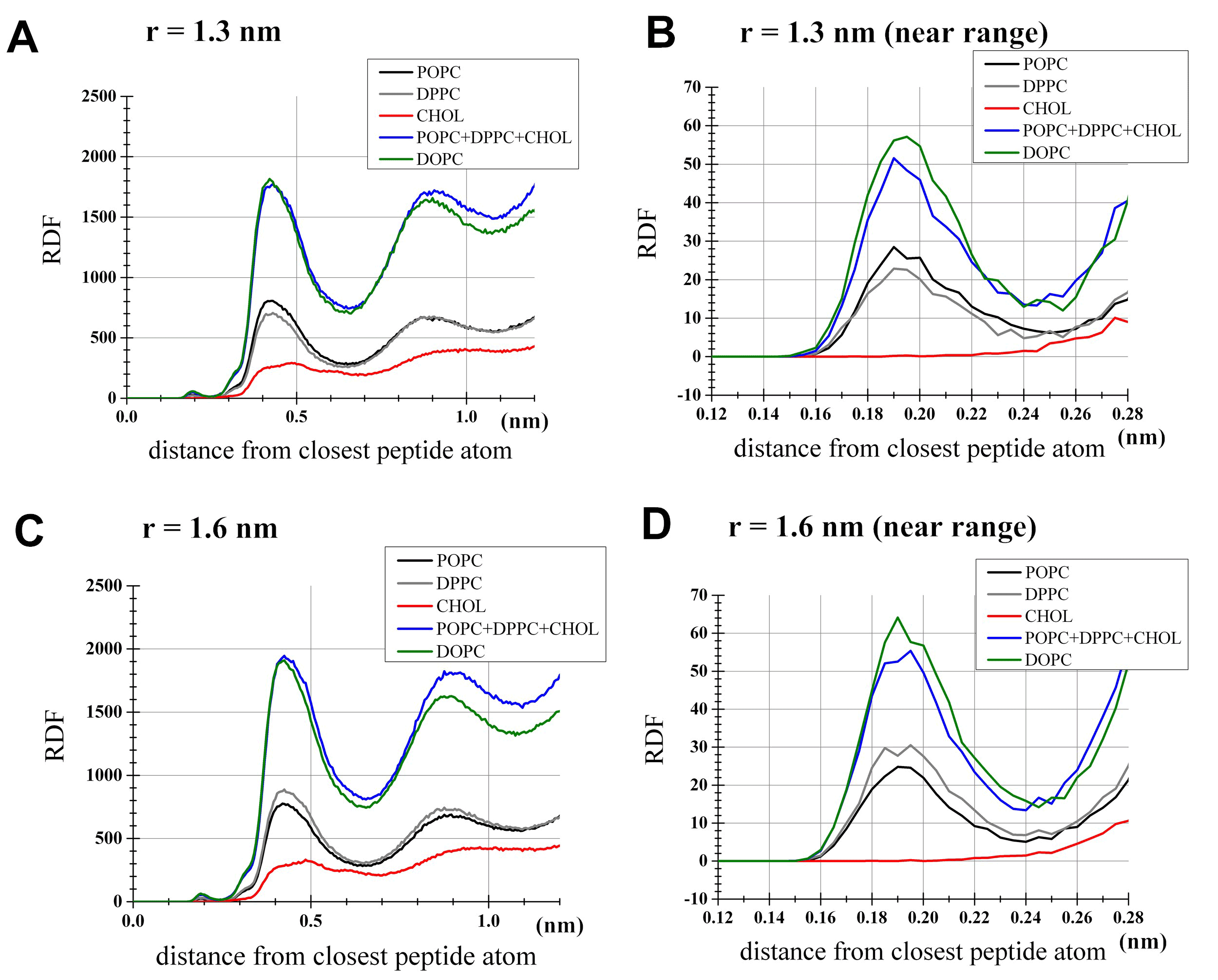
Figure 3. Radial distribution function (rdf) analysis for lipids. For each lipid species shown is the unnormalized density profile of lipid atoms located at indicated distances from the nearest atom of the peptides. The profile for DOPC was based on the DOPC set of the PMF analysis, while the POPC, DPPC and cholesterol profiles were from the 1:1:1 set. (a) Simulations with the restraint of interhelical distance at r=1.3 nm as an example for the dimerized state. Shown by a blue line is the sum of POPC, DPPC and cholesterol densities in the 1:1:1 umbrella simulation. (b) Same as (a), but the near range (<0.28 nm) was highlighted for clarity. (c) Simulations with r=1.6 nm, representing the monomers state. (d) Same as (c) but with the near range highlighted.
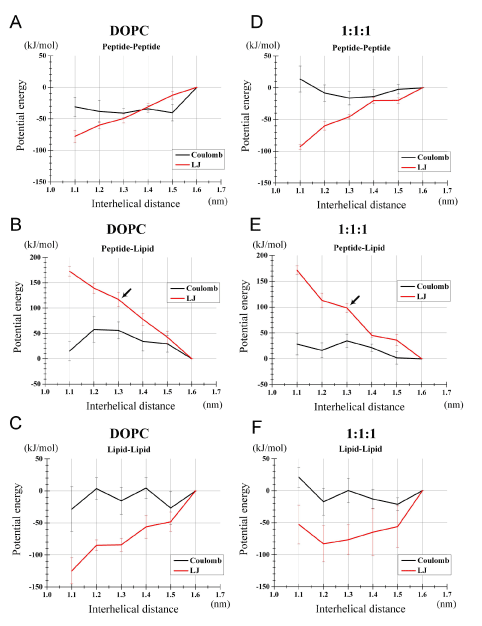
Figure 4. Decomposition of the potential energies (i.e., Coulombic and LJ energies) into lipid-lipid, peptide-lipid and peptide-peptide interaction energies. Results based on the simulations of the PMF analysis are shown: (a-c) for the DOPC bilayer and (d-f) for the 1:1:1 bilayer. (a, d) Specific peptide-peptide potential energy. (b, e) Specific peptide-lipid potential energy. (c, f) Specific lipid-lipid potential energy.
The lipid-peptide potential energy term is important in the dimer stabilization, suggesting a relatively uncompromised solvation of dimer peptides by lipids in the raft-like bilayer
Is the high dimerization propensity in the DPPC- and cholesterol-rich bilayer (Figure 2) caused by some factors that lowers the potential energy of protein-protein interaction? Consideration of thermodynamic parameters becomes important in such discussions. In theory, the free energy profiles can be decomposed into enthalpy and entropy components [34]. ΔGdim was derived from the PMF profile as we have seen above. The enthalpy change ΔHdim was inferred from the total energy of the system, ignoring the pressure-volume term. The entropy component TΔSdim was inferred from the relationship of ΔGdim =ΔHdim - TΔSdim. Here we acknowledge that possible effect of the umbrella potential that could modify the total energy possibly causing between-window differences were ignored. In this study we estimate the ΔHdim value using the values at r=1.3 and 1.6 nm. Particular errors could arise from truncation at 1.6 nm and call for a critical assessment in future studies. From our drawing the curves, ΔHdim (±SE) was estimated to be 3 (±22) kJ/mol and -TΔSdim to be -3 (±22) kJ/mol upon the peptides approach from 1.6 nm to 1.3 nm position in the DOPC bilayer (details not shown). For the 1:1:1 bilayer, the corresponding values were ΔHdim=-25 (±41) kJ/mol and -TΔSdim=23 (±41) kJ/mol. Recently, Yano et al. [35] showed that dimerization of (AALALAA)3 in a POPC membrane is an enthalpy-driven process (ΔGdim=-13.2 kJ/mol, ΔHdim=-23.7 kJ/mol, and -TΔSdim=10.4 kJ/mol) and that cholesterol addition to the POPC bilayer increased the unsigned value of ΔGdim and ΔHdim thus stabilizing the peptide dimerization (ΔGdim=-22.6 kJ/mol, ΔHdim=-84.1 kJ/mol, and -TΔSdim=61.4 kJ/mol). Thus, although our limited lengths of computation led to large SE values, our results were consistent with the experimental conclusion that the dimerization is an enthalpy-driven process and showed that cholesterol and saturated FA chains strengthened the dimer-stabilizing effect through increasing the unsigned value of the enthalpy. The discrepancies between our computation and the experiment are likely to be caused by many factors involving enthalpy-entropy compensation in UA model, differences in lipid composition and definition of dimerization, and inaccuracies of parameters.
In general, dimerization of TM peptides is mainly governed by three components of the total energy that approximate the enthalpy: peptide-peptide, peptide-lipid, and lipid-lipid interaction potential energies. If the peptide-lipid interaction component is weak or not-attractive, then peptides can be easily desolvated from lipids and dimerization is favored [28]. Is this the case with the dimer stabilization in the 1:1:1 bilayer? Or alternatively, does strong lipid-lipid interaction of the raft-like bilayer act to segregate the peptides, stabilizing the dimerized state? To better address these questions, we conducted a decomposition analysis of the total potential energy into three specific components: the specific potential energy of peptide-peptide interaction Vpept-pept, that of peptide-lipid interaction Vpept-lipid, and that of lipid-lipid interaction Vlipid-lipid. We, for example, let Vpept-pept (r) denote the peptide-peptide potential energy when the interhelical distance was r nm. This can be further decomposed into LJ energy VLJpept-pept (r) and Coulombic energy term VCoulpept-pept (r). Here again, following Castillo et al. [34], this decomposition dealt with the potential energy (the sum of the LJ and electrostatic energies), ignoring the umbrella potential and the pressure-volume term, which is usually negligibly small [34].
Compared to the monomers state (r =1.6 nm), the dimerized peptides (r=1.3 nm) showed a more negative (favorable) peptide-peptide interaction potential energy (Figures 4A, 4D), that is, Vpept-pept (1.3) < Vpept-pept (1.6), for both LJ and Coulombic energies. This is not surprising as both the LJ and electrostatic energy functions basically have such a property. It was also noted that the differential {VLJpept-pept (1.3) - VLJpept-pept (1.6)} was similar between the two bilayers (Figures 4A, 4D), as the differential is solely dependent on structures and positions of peptides. {VCoulpept-pept (1.3) - VCoulpept-pept (1.6)} was more negative for the DOPC than for the 1:1:1 bilayer, but this Coulombic term did not explain the increased stabilization of the peptide dimer in the 1:1:1 bilayer.
More importantly, the specific peptide-lipid interaction energy VLJpept-lipid at r=1.3 nm was greater than at r=1.6 nm by 117 kJ/mol for the DOPC bilayer, indicating that peptide dimerization was accompanied by unfavorable or sparse (poorly fitting) interaction between peptides and lipids (Figure 4B, black arrow). Intuitively, this implies that peptide dimerization expels out many of lipid molecules that would have been in contact with peptides placed at a large interhelical separation. Strikingly, VLJpept-lipid (1.3) was 'to a lesser extent' higher than VLJpept-lipid (1.6) in the 1:1:1 bilayer relative to the DOPC bilayer, with the differential being 100 kJ/mol (Figures 4B, 4E, black arrows). Of note, this may be a conservative interpretation; because the PMF profile (y-axis of the Figure 2) normally decreases with r growth due to the space increase, and also based on the results shown in Figure 2, the mean of VLJpept-lipid (1.3) and VLJpept-lipid (1.2) could better represent the value of the dimerized state and, accordingly, the difference between the two bilayers in VLJpept-lipid (r) should be even clearer. Intriguingly, VCoulpept-lipid (r) term also contributed to the dimer stabilization in the 1:1:1 as opposed to the DOPC bilayer (Figures 4B, 4E) despite that only the backbone atoms of the peptides and atoms of lipid headgroups are contributing to this term. This implies that the 1:1:1 bilayer lipids can solvate (fit) the peptides better even after the peptides dimerize compared to the DOPC bilayer, when the monomers state is used as the reference.
Above findings about the peptides segregation from cholesterol (Figure 3) led us to anticipate that the lipid-lipid potential Vlipid-lipid could also be an important factor stabilizing the peptide dimer. However, the differential {VLJlipid-lipid (1.3) - VLJlipid-lipid (1.6)} was -84 kJ/mol for the DOPC and -78 kJ/mol for the 1:1:1 bilayer (Figures 4C, 4F), arguing against the role of this term for the dimer stabilization in the 1:1:1 bilayer. The corresponding profiles of VCoullipid-lipid did not explain the dimer stabilization in the 1:1:1 bilayer, either (Figure 4C, 4F). Thus, for both bilayers, the lipid-lipid component showed peptide dimer-stabilizing effects, but this effect was slightly greater for the DOPC than for the 1:1:1 bilayer. Thus, contrary to our anticipation that the raft-like 1:1:1 bilayer would have tight lipid-lipid interactions that segregate the peptides, the lipid-lipid interaction potential energy could not explain the high dimerization propensity in the 1:1:1 bilayer.
Taken together, these findings indicate that the 1:1:1 bilayer, which was rich in saturated FA chains of phospholipids and cholesterol, stabilized the self-dimerization of a helical TM poly-Ile peptide compared to the DOPC bilayer. This difference was mainly accounted for by the better solvation of peptides by phospholipids in the 1:1:1 bilayer relative to the DOPC bilayer when a peptide dimer was formed. Expulsion of peptides from phospholipids/cholesterol aggregates may have a supportive role in the dimer stabilization, but our potential energy decomposition analysis did not support its importance for the dimer stabilization. Rather, some structural features, possibly highly ordered acyl chains, of POPC and DPPC molecules surrounded by cholesterol-rich sub-compartments may confer these phospholipids the ability to better solvate dimerized peptides. In the DOPC bilayer, a lower order of lipids acyl chains may make peptide-lipid contact non-optimum when the peptide dimerize, thereby increasing the energy cost for peptide dimerization.
Discussion
In this study, a bilayer with a cholesterol- and saturated FA-rich composition stabilized dimerization (dimerization energy of -2.23 kJ/mol) compared to the DOPC bilayer (-0.01 kJ/mol), for a TM helical peptide with a sequence consisting only of 21 Ile residues. The potential energy decomposition showed that, upon dimerization, the peptide-lipid interaction energy becomes high (unfavorable) (Figures 4B, 4E). This was understandable as dimerization of peptides more or less expels lipids, partially hindering contact (solvation) of lipids to peptides. Intriguingly, for both LJ and Coulombic energies, the energy cost for peptide dimerization due to the peptide-lipid interaction potential energy term (i.e., the cost due to peptide desolvation from lipids) was not so high for the 1:1:1 bilayer compared to the case with the DOPC bilayer (Figures 4B, 4E). This implies that, even after peptides dimerize, some solvation (fitting) of peptides by lipids is possible in the 1:1:1 bilayer, mitigating the cost due to the desolvation upon dimerization. Contrary to our anticipation, the profile of the lipid-lipid potential energy did not explain the high dimerization propensity for the 1:1:1 bilayer (Figures 4C, 4E). Thus, although segregation of peptides from the cholesterol-rich sub-areas was observed, it was unlikely that the thermodynamically-favored formation of phospholipids/cholesterol aggregates directly augments the peptide dimerization propensity. Rather, phospholipids/cholesterol aggregates augments the lipid order parameters of the system, and this may provide some advantages in the solvation after peptide dimerization, reducing the energy cost for desolvation of peptides from lipids. Although further analysis is necessary to figure out the advantages, it is possible that straightened lipid chains (high order parameters) may directly improve solvation of peptides.
Is this dimer-stabilizing effect of saturated FA and cholesterol specific to the poly-Ile peptide or common to many helical TM peptides? The computational burden prevented us from testing many peptides, but, when we tested a TM helix with a sequence of I(VI)10, (i.e., 21-residue peptide with alternating Ile and Val) with 300 ns for each window, it also showed similar dimer stabilization by -1.8 kJ/mol in the 1:1:1 relative to the DOPC bilayer. The decomposition analysis of the I(VI)10 peptide showed a similar trend to the poly-Ile, with {VLJpept-lipid (1.3) - VLJpept-lipid (1.6)} being 110 kJ/mol for the DOPC and 98 kJ/mol for the 1:1:1 bilayer (Nishizawa unpublished data). This differential showed even greater difference between the two bilayers when VLJpept-lipid (1.2) was used instead of VLJpept-lipid (1.3). The differential in the Coulombic energy {VCoulpept-lipid (1.3) - VCoulpept-lipid (1.6)} was also lower for the 1:1:1 (25 kJ/mol) relative to the DOPC bilayer (60 kJ/mol), suggesting a contribution of this term to the dimer stability. For the I(VI)10 peptide as well, Vlipid-lipid did not explain the dimerization-stabilizing effect of the 1:1:1 relative to the DOPC bilayer. Thus, in both the PMF analysis and the potential energy decomposition analysis, I(VI)10 showed a similar feature to the poly-Ile. We speculate that most helical TM peptides show the trend that the saturated FA and cholesterol decrease the energy cost of desolvation via relatively favorable lipid-protein interaction after dimerization, leading to the dimer-stabilizing effect of these lipids.
Dimerization/oligomerization of multi-spanning receptor proteins has also been recognized as part of normal signaling process, as seen in LH receptors (see Introduction). As another example, Hwang and coworkers showed that saturated FAs activated TLR4, but DHA inhibits lipopolysaccharide (LPS)-induced activation of TLR4, and that the molecular target for this phenomena being receptor itself or the upstream events leading to TLR4 activation [36]. More recently Wong et al. used macrophage RAW264.7 cells and showed that lauric acids, but not DHA, facilitate association of TLR4 and its adaptor molecules with lipid rafts (based on sucrose gradient fractionation), also facilitating dimerization and activation of TLR4 [37]. Thus, oligomerization status is likely to be a factor controlling partition of receptor proteins into lipid rafts [21].
However, there are non-specific and specific effects of lipid on protein behaviors confounding each other, making analysis and interpretation difficult. Specific interactions are likely to help recruitment proteins to particular microdomains, fine-tuning some cellular signaling. However, except for FA derivatization (modification), clear recruitment signals to such microdomain have not been well known [15,16]. In living cells raft and non-raft microdomains have rather small differences, raising a possibility that effects of FA and cholesterols on protein dynamics are at least in part, not dependent on the presence of a specific types of microdomains. In physiological systems, cholesterol is contained not only in raft but also in non-raft domains. Analyses of diverse compositions may provide further insights into the question whether the observed finding is relevant to a wide range of bilayer including both raft and non-raft domains. In this study, we chose extreme compositions, i.e., a 1:1:1 POPC/DPPC/cholesterol bilayer and a pure DOPC bilayer, but many more compositions may be worth analysis. Although a limited level of resolution hampered discussion of relative importance of saturated FA and cholesterol in this study, varying composition focusing on each of FA and cholesterol would be informative. This line of extension of this project is underway.
References
- Calder PC (2008) The relationship between the fatty acid composition of immune cells and their function. Prostaglandins Leukot Essent Fatty Acids 79: 101-108. [Crossref]
- Estadella D, da Penha Oller do Nascimento CM, Oyama LM, Ribeiro EB, Dâmaso AR, et al. (2013) Lipotoxicity: effects of dietary saturated and transfatty acids. Mediators inflamm 2013: 137579. [Crossref]
- Endres S, Meydani SN, Ghorbani R, Schindler R, Dinarello CA (1993) Dietary supplementation with n-3 fatty acids suppresses interleukin-2 production and mononuclear cell proliferation. J Leukoc Biol 54: 599-603. [Crossref]
- Kelley DS, Taylor PC, Nelson GJ, Schmidt PC, Ferretti A, et al. (1999) Docosahexaenoic acid ingestion inhibits natural killer cell activity and production of inflammatory mediators in young healthy men. Lipids 34: 317-324. [Crossref]
- Calder PC, Bond JA, Bevan SJ, Hunt SV, Newsholme EA (1991) Effect of fatty acids on the proliferation of concanavalin A-stimulated rat lymph node lymphocytes. Int J Biochem 23: 579-588. [Crossref]
- Calder PC, Yaqoob P, Harvey DJ, Watts A, Newsholme EA (1994) Incorporation of fatty acids by concanavalin A-stimulated lymphocytes and the effect on fatty acid composition and membrane fluidity. Biochem J 300: 509-518. [Crossref]
- Ruiz-Núñez B, Dijck-Brouwer DA2, Muskiet FA (2016) The relation of saturated fatty acids with low-grade inflammation and cardiovascular disease. J Nutr Biochem 36: 1-20. [Crossref]
- Fernanda Cury-Boaventura M, Cristine Kanunfre C, Gorjão R, Martins de Lima T, Curi R (2006) Mechanisms involved in Jurkat cell death induced by oleic and linoleic acids. Clin Nutr 25: 1004-1014. [Crossref]
- Shaikh SR, Boyle S, Edidin M (2015) A high fat diet containing saturated but not unsaturated fatty acids enhances T cell receptor clustering on the nanoscale. Prostaglandins Leukot Essent Fatty Acids100: 1-4. [Crossref]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, et al. (2011) Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147: 173-184. [Crossref]
- Solinas G, Naugler W, Galimi F, Lee MS, Karin M (2006) Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. 103: 16454-16459. [Crossref]
- Calder PC (2013) n-3 fatty acids, inflammation and immunity: new mechanisms to explain old actions. Proc Nutr Soc 72: 326-336. [Crossref]
2021 Copyright OAT. All rights reserv
- de Jong AJ, Kloppenburg M, Toes RE, Ioan-Facsinay A (2014) Fatty acids, lipid mediators, and T-cell function. Front Immunol 5: 483. [Crossref]
- Hubler MJ, Kennedy AJ (2016) Role of lipids in the metabolism and activation of immune cells. J Nutr Biochem 34: 1-7. [Crossref]
- Stulnig TM, Zeyda M (2004) Immunomodulation by polyunsaturated fatty acids: impact on T-cell signaling. Lipids 39: 1171-1175. [Crossref]
- Lorent JH, Levental I (2015) Structural determinants of protein partitioning into ordered membrane domains and lipid rafts. Chem Phys Lipids 192: 23-32. [Crossref]
- Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, et al. (2005) Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11: 90-94. [Crossref]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, et al. (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687-698. [Crossref]
- Lingwood D, Simons K (2010) Lipid rafts as a membrane-organizing principle. Science 327: 46-50. [Crossref]
- Levental I1, Grzybek M, Simons K (2011) Raft domains of variable properties and compositions in plasma membrane vesicles. Proc Natl Acad Sci U S A 108: 11411-11416. [Crossref]
- Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K (2015) Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 72: 557-581. [Crossref]
- Stulnig TM, Huber J, Leitinger N, Imre EM, Angelisova P, et al. (2001) Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem 276: 37335-37340. [Crossref]
- Moore DT, Berger BW, DeGrado WF (2008) Protein-Protein Interactions in the Membrane: Sequence, Structural, and Biological Motifs. Structure 16: 991-1001. [Crossref]
- Roess DA, Smith SM (2003) Self-association and raft localization of functional luteinizing hormone receptors. Biol Reprod 69: 1765-1770. [Crossref]
- Smith SM, Lei Y, Liu J, Cahill ME, Hagen GM, et al. (2006) Luteinizing hormone receptors translocate to plasma membrane microdomains after binding of human chorionic gonadotropin. Endocrinology 147: 1789-1795. [Crossref]
- Kaiser HJ, Lingwood D, Levental I, Sampaio JL, Kalvodova L, et al. (2009) Order of lipid phases in model and plasma membranes. Proc Natl Acad Sci U S A 106: 16645-16650. [Crossref]
- Nishizawa M, Nishizawa K (2014) Potential of mean force analysis of the self-association of leucine-rich transmembrane a-helices: difference between atomistic and coarse-grained simulations. J Chem Phys 141: 075101. [Crossref]
- Nishizawa M, Nishizawa K (2016) Free energy of helical transmembrane peptide dimerization in OPLS-AA/Berger force field simulations: inaccuracy and implications for partner-specific Lennard-Jones parameters between peptides and lipids. Molecular Simulation 42: 916-926.
- Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 4: 435-447. [Crossref]
- Malde AK, Zuo L, Breeze M, Stroet M, Poger D, et al. (2011) An Automated force field Topology Builder (ATB) and repository: version 1.0. J Chem Theory Comput 7: 4026-4037. [Crossref]
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem 13: 1011-1021.
- Niemelä PS, Ollila S, Hyvönen MT, Karttunen M, Vattulainen I (2007) Assessing the nature of lipid raft membranes. PLoS Comput Biol 3: e34. [Crossref]
- Veatch SL, Keller SL (2005) Seeing spots: complex phase behavior in simple membranes. Biochim Biophys Acta 1746: 172-185. [Crossref]
- Castillo N, Monticelli L, Barnoud J, Tieleman DP (2013) Free energy of WALP23 dimer association in DMPC, DPPC, and DOPC bilayers. Chem Phys Lipids 169: 95-105. [Crossref]
- Yano Y, Kondo K, Kitani R, Yamamoto A, Matsuzaki K (2015) Cholesterol-induced lipophobic interaction between transmembrane helices using ensemble and single-molecule fluorescence resonance energy transfer. Biochemistry 54: 1371-1379. [Crossref]
- Lee JY, Ye J, Gao Z, Youn HS, Lee WH, et al. (2003) Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem 278: 37041-37051. [Crossref]
- Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, et al. (2009) Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem 284: 27384-27392. [Crossref]