Cancer is a general term to define a group of genetic alterations and the subsequent phenotypic and physiological changes at the cellular level, leading to accelerated death of the host. According to The World Health Organisation (WHO), it is the second leading factor for human deaths globally. Cancer is such a difficult challenge for mankind because of its ability to relapse even after detailed treatments with chemotherapeutic agents and ionizing radiations. This can be explained by the emerging concept of ‘tumor initiating cells’ or ‘cancer stem cells’ (CSCs) which comprise a pluripotent and genomically dynamic subpopulation of the tumor microenvironment, occupying only 0.1%-3% of the total tumor population. Despite their emerging significance in disease progression and recurrence, the origin of these CSCs and their complex interaction with the corresponding CSC niche still remain to be elusive. In this review, we make an attempt to unwind the mystery of the origin of the CSCs as well as focus on the well-established cross-talk mechanisms between the CSCs and their niche components. Lastly, we try to find alternative therapeutic strategies to combat CSC resistance by targeting the various components of the CSC niche.
cancer stem cells, cancer stem cell niche, cancer stem cell origin, niche targeted therapy, VSELs
Abbreviations
ABC: ATP-binding cassette, bFGF: Basic fibroblast growth factor, BCL-XL: B-cell lymphoma-extra-large, BMI1: B lymphoma Mo-MLV insertion region 1, CAFs: Cancer associated fibroblasts, CSCs: Cancer stem cells, CXCR: CXC Chemokine receptor, EMT: Epithelial to mesenchymal transition, ECM: Extracellular matrix, IL: Interleukin, KLF4: kruppel like factor 4, LIF-JAK-STAT: Leukemia inhibitory factor-janus kinase-signal transducer and activator of transcription, MDR: Multidrug resistance, MRP1: Multidrug resistance associated protein 1, MSCs: Mesenchymal stem cells, NSCCs: Non-stem cancer cells, OCT4: Octamer-binding transcription factor 4, SHH: Sonic hedgehog,SOX2: Sex determining region Y box-2, TAFs: Tumor associated fibroblasts, TAMs: Tumor associated macrophages, TANs: Tumor associated neutrophils, TGF-β: Transforming growth factor beta,TNF-α: Tumor necrosis factor-α, VEGF: Vascular endothelial growth factor
Cancer is one of the most life-threatening diseases in today’s world [1]. The advancement in medical science has failed to find out a proper cure for cancer. Chemotherapy and radiotherapy may reduce the cancer burden but fail to completely eradicate the disease [2]. Studies done by various groups have identified a rare sub-population of cell present within the tumor mass which they termed as tumor initiating cell or cancer stem cells (CSCs) [3].
Further research on CSCs showed that these cells have properties similar to pluripotent stem cells such as self-renewal, quiescence and ability to differentiate into any lineage specific cell type [4]. Several reports have also suggested the existence of certain common gene signatures, biomarkers, epigenetic regulators and signalling pathways between pluripotent stem cells and CSCs [5]. Self-renewal is a crucial property of all stem cells through which they maintain a relative equilibrium between self-regeneration and differentiation. Many of the signalling pathways that modulate normal stem-cell self-renewal are dysregulated in CSCs, thereby leading to aberrant or unregulated self-renewal and neoplastic growth of CSCs [6]. Self-renewal of CSCs is achieved through a process of symmetric cell division via upregulation of Human telomerase reverse transcriptase (hTERT), WNT3a, HEDGEHOG, etc. [7-8]. Commonly stem cells are considered to be quiescent in nature. However, in some instances these stem cells are activated and they enter into cell cycle from time to time to maintain homeostasis or a steady state. Bradford et al. [9] showed the cell cycling characteristics of hematopoietic stem cells (HSCs), thereby confirming that the stem cells are not dormant, but they move through the cell cycle in a slower rate to maintain a normal steady-state bone marrow. Similarly, according to Glauche et al. [10] HSCs can reversibly switch between an activated and a quiescent state, CSCs also have analogous property which helps them to shield themselves from radiotherapy and chemotherapy and also establish secondary metastasis [11]. There is a long list of evidence which indicates that CSCs could differentiate into any cell type, such as neuroendocrine cells or endothelial-like cells, in response to appropriate cues for differentiation [12].
CSCs have now gained prime focus in the area of cancer research and have been reported in most human tumors. CSCs are commonly identified from haematological and solid tumors using markers specific for normal stem cells such as CD133 [13] (also known as Prominin1), CD24 [14], CD44 [15], ABCG2 [16], Thymocyte differentiation antigen 1 (THY1) [17], Aldehyde dehydrogenase (ALDH) [18] etc. Subsequently, various novel markers such as CD47 [19], CD96 [20], T-cell immunoglobulin and mucin-domain containing-3 (TIM3) [21], C-type lectin-like molecule-1 (CLL-1) [22], CD25 and CD32 [23] too have a high expression on leukemia stem cells than normal hematopoietic stem cells. These surface markers are often used in conjugation with each other for various types of tumors. Some of the key transcription factors like Sex determining region Y box-2 (SOX2), Octamer-binding transcription factor 4 (OCT4), kruppel like factor 4 (KLF4), NANOG and cMYC that regulate the pluripotency, are also over-expressed in CSC population [24].
Tumor initiation
CSCs or tumor initiating cellshave the potential to repopulate the entire tumor mass [25]. A study by Boumahdi et al. [26] reported that SOX2 upregulation in CSCs plays a critical role in associating the entire process of tumor initiation, progression and maintenance. Li et al. [27] have shown as to how CD54 over-expressed cancer cell can initiate tumor formation by p38-regulated NOTCH1 signalling. Several reports indicated the existence of a distinct cell surface marker specific subpopulation within a heterogeneous tumor such as CD34+/CD38- (Leukemia) [28], CD133+/CD44+ (colon cancer) [29], CD44+/CD24-(breast cancer) [15] etc., that can induce tumor formation in both mice and humans.
Angiogenesis
The above-mentioned CSC-enriched subpopulation, being extraordinarily tumorigenic, release plentiful of Vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), IL-6, IL-8and angiogenin to stimulate the process of tumor-angiogenesis [30]. Microarray analyses exhibited elevated levels of VEGF, angiopoietin 2 and hypoxia inducible 1α factor (HIF1α) from glioma cell neurosphere culture and reduced levels of thrombospondin 1(angiogenesis inhibitor) as compared to monolayer culture of the same [31]. Related research in breast cancer disclosed over-expression of VEGF in CSC-enriched MCF-7 sphere cultures as matched to CSC-deprived monolayer MCF-7 culture [32].
Metastasis
Establishment of angiogenesis is subsequently followed by distant metastasis. In the entire process of tumorigenesis, the epithelial-to-mesenchymal transition (EMT) plays a very significant role [33]. A recent study by Mukherjee et al. [34] classified cancer stem cells as intrinsic CSCs and metastatic CSCs on the basis of their role in tumor metastasis. Intrinsic CSCs, which reside in the inner core of the tumor, remain passive during the process of metastasis but help in generation of CXCR4-expressing metastatic CSCs from non-stem cancer cells via paracrine signalling. These actively participating metastatic CSCs are found in the peripheral/disseminating region of the tumor [34]. Certain genes which are associated with the mesenchymal phenotype such as TWIST, SNAIL, SLUG, VIMENTIN, HZEB1 etc., are upregulated during EMT in metastatic CSCs [35-37].
Drug resistance and Radio-resistance
Recent reports support a view that these CSCs are also responsible for drug-resistance and relapse [38]. It has been well documented that the multidrug resistance (MDR) of the CSC population is conferred by the over-expression of P-glycoproteins and/or ABC transporters (drug-efflux pumps) such as ABCC1/MRP1, ABCB1 and ABCG2 [39-40]. Also, the radio-resistance of CSCs is mediated by stimulating the DNA damage checkpoint response upon exposure to harmful radiations that involves preferential activation of ataxia telangiectasia mutated (ATM) protein kinase along with checkpoint kinases (Chk1 and Chk2) [41]. The quiescent nature of CSCs shields them from various apoptotic signals, anti-proliferative agents, and makes them insensitive towards any conventional chemotherapy [42] (Figure 1).
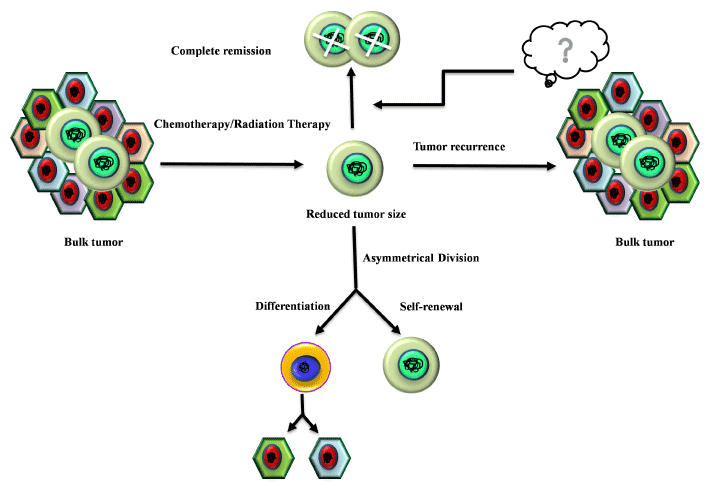
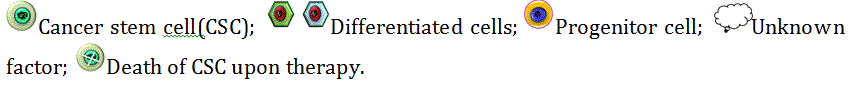
Figure 1. Chemoresistance and radioresistance property of the CSCs. Chemotherapy and radiotherapy can eliminate the tumor cells but CSCs present within the tumor bulk are resistant towards both chemotherapy and radiotherapy. After surviving chemo and radiotherapy CSCs are self-sufficient to repopulate the entire tumor through renewal self-renewal and differentiation.
Above discussion highlighting the role of CSCs as an engine of tumor evolution, tempted us to explore the origin of these stem-like cells as well as the CSC niche to unveil the missing link in decoding the cancer stem cell code and to target CSCs.
The origin of CSCs has become quite a puzzle as multiple hypotheses concerning the origin of CSCs have emerged. One of those states that CSCs have emerged from the existing population of non-stem cancer cells (NSCCs) within the tumor mass. NSCCs have only a restricted proliferative capacity but constitute the bulk of the tumor. These non-stem cells, by accumulating various kinds of mutation, might gain stemness potential [43,44]. The second hypothesis explains the origin of CSCs from somatic stem cells. These stem cells are a compelling target as they survive long enough and accumulate DNA damage which might make them malignant. Somatic stem cells are usually multipotent (or sometimes unipotent), which explains the heterogeneity in most tumors. Although somatic stem cells are generally quiescent, they have remarkable self-renewal potential, which is crucial for tumor expansion [45]. The very small embryonic like stem cells (VSELs), which have been recently identified have characteristics similar to that of pluripotent stem cells and thus is considered to be another possible origin of the CSCs.
Non-stem cancer cells
According to Yanger et al. [46], CSCs arise from non-stem cancer cells by the upregulation of several pluripotency-related pathways like JAGGED2-NOTCH, WNT/β-CATENIN and Hedgehog. Similarly, inactivation of the Hippo pathway, a regulator of cell proliferation, has been noted to be sufficient to de-differentiate mature cells into cells with CSC like phenotype [47]. Interestingly, CSC-shed IL6 has also been reported to impart stemness in non-stem cancer cells [48]. Recent research implies that CSCs can also be generated from cancer cells through epigenetic modification of differentiation-related genes and/or tumor suppressor genes [49]. It has been identified that factors of polycomb repressive complex 2 (PRC2), like enhancer of zeste homolog 2 (EZH2) and suppressor of zeste 12 homolog (SUZ12), were overexpressed in various cancers like ovarian, breast, prostate, and colon cancers, and they were crucial for maintenance of the CSC population [50]. Endothelial cells, on the other hand, have also been found to enhance the stemness properties in cancer cells by upregulation of nitric oxide (NO), Sonic Hedgehog (SHH) and NOTCH signalling pathways [51] that in turn increase a number of stemness-related factors such as B lymphoma Mo-MLV insertion region 1 (BMI1), Oligodendrocyte Lineage Transcription Factor 2 (OLIG2), SOX2 and OCT4, generate CSCs [52]. On the other hand, the rapid growth rate of cancer cells results in hypoxia that upregulates many hypoxia-related genes such as HIF1α and HIF2α, which further induces dedifferentiation of non-stem cancer cells via upregulation of NOTCH-1, c-kit, EMT-specific genes like SNAIL, SLUG, TWIST, and N-cadherin, thereby causing acquisition of stem cell-like properties and metastasis [53,54].
Somatic stem cells
Another hypothesis involves the existing pool of somatic stem cells as the origin of CSCs. Under normal physiological conditions, stem cells (i) self-renew and give rise to daughter stem cells, and/or (ii) differentiate to give rise to mature progeny [55]. However, dysregulation of one/more intrinsic pathways such as WNT, SHH, Phosphatase and tensin homolog (Pten), BMI1 and NOTCH in the adult stem cells, can give rise to CSCs as well [56]. Moreover, the microenvironment, in which the stem cells reside, regulates their activity via crosstalk of various signals [57]. Disruption of these complex signals affects normal stem cell homeostasis. In addition, the efficiency of stem cells declines with aging which then leads to the accumulation of nuclear as well as mitochondrial DNA damage, thereby resulting in the onset of malignancy [58]. Experiments show that expanding adipose-derived stem cells at high oxygen concentration could increase the rate of mutation during DNA replication, as occurs in neoplastic tissues [59]. Mutation and/or loss of tumor suppressors like p21 and Tp53 could also induce transformation of stem cells into CSCs [60].
Very small embryonic/epiblast-like stem cells
At this juncture, it may not be out of context to mention that the most puzzling point is that tumors mimic tissues which are early in their developmental stage and often express early developmental markers such as α-fetoprotein. Recently, a rudimentary germ line-derived VSELs have been identified, which are deposited in several organs during embryogenesis and persist into adulthood. This might be the missing link which may answer the germ line origin hypotheses of CSCs. VSELs are small (< 6μm) cells, identified as SCA-1+CXCR4+Lin−CD45− from adult murine bone marrow [61] and CD133+ CD45- LIN- from human umbilical cord blood [62]. TEM images showed a very primitive morphology with a large nuclei surrounded by a thin cytoplasmic ring, enriched in mitochondria [63]. They have a high expression of REX-1, Stage-specific embryonic antigen 1 (SSEA-1) and 4 [64] but despite being pluripotent stem cells, they exhibit down-regulated Sox2 expression [65], which may be due to the heterogeneous nature of VSELs or its epigenetic regulation that controls pluripotency and self-renewal [66]. In contrast, some groups have reported Sox2 expression in VSELs [67]. They also express other germ line markers like Mouse vasa homologue (MVH), STELLA, NOBAX, FRAGALLIS, Dead end protein homolog 1 (DND1), Histone deacetylase 6 (HDAC6) [68]. VSELs have a high telomerase activity [69] and do not express MHC I [70]. They also share similar surface antigens including CXCR4 [71] with CSCs. The resemblance between the VSELs and CSCs designate VSELS as a potential origin of CSCs. However, there is no detail report on the properties of these VSELs (Figure 2).

Figure 2. Origin of the cancer stem cells. This figure explains the three main hypotheses regarding the CSC origin. Non-stem cancer cells give rise to CSCs by upregulating the pluripotency-associated signalling pathways such as JAGGED2- NOTCH, WNT/β-CATENIN and Hedgehog while downregulating the differentiation- inducing pathways such as the Hippo pathway. Dysregulation in the regulatory pathways of the adult stem cell homeostasis also results in the conversion of these stem cells into CSCs. VSELs, being highly similar to the pluripotent stem cells, are also considered to be a potential origin of CSCs, when exposed to mutagenesis.
Targeting the somatic stem cells to reduce or remove the CSCs is not a viable option as somatic stem cells plays a key role in tissue regeneration and homeostasis [72] and targeting them will do more harm than good. Also insignificant information is present about the role of VSELs in originating the CSCs so it is not possible to therapeutically target them as a method to diminish the CSC population.
Various groups have tried and targeted the pathways in cancer cell which leads to the origination of CSCs like Notch [73], Oct4 [74], Sox2 [75], BMK [76] pathways etc and showed that shRNA or siRNA mediated knockdown induced suppression of sphere formation or increased apoptosis, but these therapies need to be properly developed to gain clinical significance [77]. Monoclonal antibodies (mAbs) such as trastuzumab (anti-HER2), rituximab (anti-CD20), bevacizumab (anti-VEGF-A) and others, have been clinically accepted as part of the combination therapy with existing chemotherapeutic drugs [78]. Nevertheless, resistance and disease relapse are observed.
Hence it seems difficult to target CSCs with strategies based on their cells of origin. This has driven several research groups to now focus on the CSC niche as the niche is a critical element that ensure CSC survival and proliferation. It thus becomes crucial to understand the role of the CSC niche in maintaining the CSCs.
Although the concept of the stem cell niche was first introduced about 39 years ago [79], strong experimental evidences confirming its existence have recently come to the limelight. The stem cell niche is a dynamic system consisting of cellular components and cell-secreted functional molecules that take part in integrating multiple external and internal signals to calibrate cell-fate and self-renewal/differentiation rate of stem cells according to the requirements of the corresponding tissue [80,81]. Both normal stem cells and the CSCs require a suitable niche in order to sustain their own pools but the niches of healthy stem cells and the CSCs differ drastically in terms of their properties and functions. The normal stem cell niche consists of normal fibroblasts, mesenchymal cells (also referred to as stromal cells), macrophages, neutrophils, and extracellular matrix (ECM) composed of fibroblast secreted collagen fibres and proteoglycans and cell-secreted soluble factors [82].
Apparently, the CSC niche has the same components, only in an aggravated state in terms of functions. This functional aggravation within the niche components is introduced by the CSCs themselves so as to mould the niche in favour of their survival.
Cellular components of the CSC niche
The CSC niche is a distinct anatomical location within the tumor microenvironment [83]. This specific residence of the CSCs engages in the complex pathways essential for the maintenance of the homeostasis of the CSC population. The several cellular and molecular components of the CSC niche come under the modulatory effects of the CSCs resulting in exasperated functional capabilities which in turn produce a propitious environment for the CSCs. Hence, we would further explore the contribution of the cellular components in the CSC niche.
- Tumor associated fibroblasts (TAFs): Tumor associated fibroblasts (TAFs) are one of the essential cellular components of the tumor niche. Kalluri et al. [84] presented that normal fibroblasts are activated into over-proliferative TAFs by the CSCs via the upregulation of WNT signalling. CSCs also secrete SHH in the niche which in turn activates the TAFs through the activation of Hedgehog signalling. As demonstrated by Valenti et al. [85] application of the Hedghog signalling inhibitor vismodegib results in reduced TAF and CSC population. Tumor associated fibroblasts (TAFs) is a chief source of IL-6 (multifunctional cytokine) and is a key component of tumor niche [86]. TAFs modulate the tumor niche by inducing angiogenesis by secreting various cytokines [87,88]. Recent reports showed the role of TAFs in drug resistance, which is classified into two different types, that is soluble factor-mediated drug resistance (SFM-DR) and cell-adhesion-mediated drug resistance (CAM-DR) [89,90]. TAFs induces EMT by secreting carbonic anhydrase IX (CA IX) which causes acidification of ECM and further enhances the activities of MMP-2 and MMP-9 [91]. TAFs mediated overexpression of BCL-xL(B-cell lymphoma – extra-large) protect cancer cells from cytotoxic effects of tyrosine kinase inhibitors [92].
- Tumor Associated Macrophages (TAMs): The primary tumor niche is defined by chronic inflammation by the NK-cells, CD8+ T cells [93,94] and its corresponding mediator, the CD103+ Dendrictic cells [95]. This chronic inflammation promotes continuous tumor proliferation and metastasis [96]. TAMs and TANs are pivotal in protection of the CSCs from the continuous immune surveillance within the tumor niche. The origin of TAMs has remained controversial [97,98] but the role of CSC-secreted Colony stimulating factor-1 (CSF-1) in TAM activation and survival is well established. CSF-1 is secreted in a high concentration by the tumor cells within the tumor niche as shown by Smith et al. [99] in endometrial cancer, and is directly correlated with TAM-associated tumor progression and metastasis as shown in the cervical, pancreas and breast models of cancer [98]. Granulocyte macrophage colony stimulating factor (GM-CSF) secreted by breast cancer stem cells also directly converts tissue macrophages into TAMs. Several researches also showed that VEGF-A overexpression by CSCs results in the conversion of macrophage progenitors into TAMs in the presence of high IL-4 concentrations in a xenograft model of skin cancer [100,101]. The Endothelial Tyrosine Kinase positive (TIE2+) TAMs present along the lining of the intra-niche vasculature direct the CSCs along the collagen network towards the region of dissemination [98].
- Tumor Associated Neutrophils (TANs): TANs are another type of polarized immune cells within the tumor niche [95]. CSCs recruit TGF-β in order to polarize the neutrophils into pro-tumorTANs [102]. Inhibition of TGF-β by SM-16 has been shown to result in the loss of pro-tumor phenotype of the TANs and the subsequent gain of anti-tumor phenotype as demonstrated by Fridlender et al. [103]. Another mechanism of CSC mediated TAN recruitment is through the surface receptors of TANs, CXCR1 and CXCR2. Raccosta et al. [104] reported that CSC-secreted liver X receptor ligand oxysterol (LXR) directly converts neutrophils into TANs through binding to CXCR2. MDSCs have also been demonstrated to give rise to TANs under the influence of TGF-β. Cytokines like CXCL-5 and TNF-α have also been reported to recruit TANs within the tumor niche [105,106]
- T-regulatory cells (Treg cells): The Treg cells limit pathogenic immune responses towards self-antigens and foreign antigens. These are FOXP3+ cells which have recently been implicated in progression of tumors (Figure 3). The Treg cells help in immune evasion of CSCs and many publications have highlighted the role of TAMs and TAFs in recruiting the Treg cells in the tumor microenvironment [107]. Recently Zhu et al. [108] showed that TAMs and Treg cells promote ovarian cancer proliferation, metastasis and tumor angiogenesis. Moreover, over-expression of VEGF by the Treg cells leads to the promotion of tumor angiogenesis [109]. TANs also participate in the recruitment of Treg cells through the secretion of CCL17 [110].
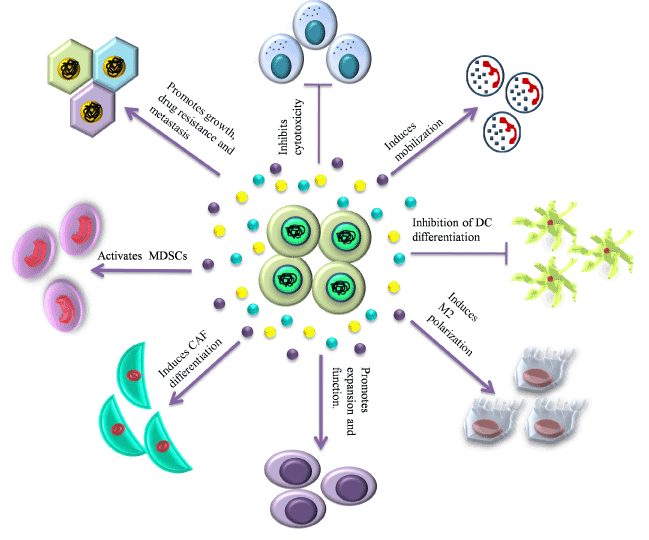
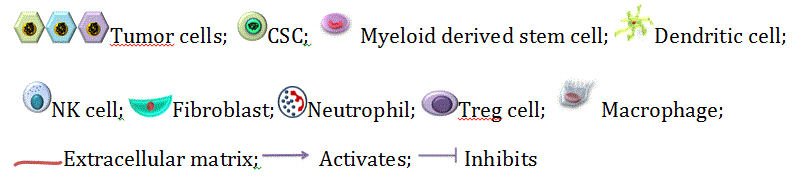
Figure 3. CSC niche: the perfect ‘home’ for CSCs. This figure depicts the interaction between the CSCs and the various cellular and soluble components of the CSC niche.
- Mesenchymal stem cells (MSCs): MSCs, another vital component of the primary tumor niche, maintain the CSC population by the activation of NK-kB pathway via secretion of CXCL12, IL-6, IL-8 [96] and over-expression of miR199a [111]. Several reports demonstrated cytokine-mediated crosstalk between MSCs and cancer cells affects the tumor niche by enhancing the generation of CSCs and their maintenance [112]. MSCs further support the tumor microenvironment by differentiating into TAFs [113].
- Cell-mediated Adhesion: Adhesion of the CSCs to the ECM and other cellular components of the niche is pivotal for stemness retention and tumor progression; stemness promoting signalling such as Hedgehog and NOTCH signalling pathways require direct cell to cell contact for receptor-ligand interactions as shown by Gilbertson and Rich [114]. Disseminated tumor cells (DTCs) adhere themselves with circulating platelets in order to prevent damage from NK cell mediated lysis, shear forces of the blood flow and to establish better contact with the endothelium of the metastatic niche [115].
Soluble-factors of the CSC Niche
The functional soluble factors within the tumor niche mainly includes crucial ECM proteins which take part in cell-cell and cell-matrix adhesion, pro-tumor cytokines, growth factors and cell-secreted micro-particles or exosomes which function as molecular vehicles for cell to cell communication. The key ECM proteins like tenascin-C expressed by the TAFs modulate tumor invasiveness. Tenascin-C contributes to the pro-invasive environment of the colon CSCs in a RhoA and Rac-dependent pathway [116].
- Secreted by cells of the niche: Cabarcas et al. [96] showed in a study that TAF-secreted MMPs-2, 3 and 9 remodel the ECM in order to promote EMT and the consequent dissemination of the cancer stem cells, a prerequisite in the process of metastasis (Figure 4) [96,117]. MMP-3 and HGF (hepatocyte growth factor) secreted by the TAFs, stimulates WNT signalling and the consequent restoration of stemness in the non-stem tumor cells [83]. Excessive secretion of type-I collagens and proteoglycans from the TAF also forms a protective barrier around the CSCs thereby preventing chemotherapeutic drugs from reaching the CSC population [118], thus contributing to drug resistance. VEFG, PDGF, TGF-β and the stemness-favoring cytokines, IL-6, IL-8 and CXCL12 secreted by the TAFs, TAMs, TANs and MSCs are the pivotal functional molecules that modulate the tumor niche in favour of CSC proliferation and invasion [83]. BMP (bone morphogenic protein) and Gremlin 1, the antagonist of BMP, are two signalling molecules highly expressed within the tumor niche [119]. Also, the differentiated glioblastoma cells secrete IL-6 which maintains CSCs [119].
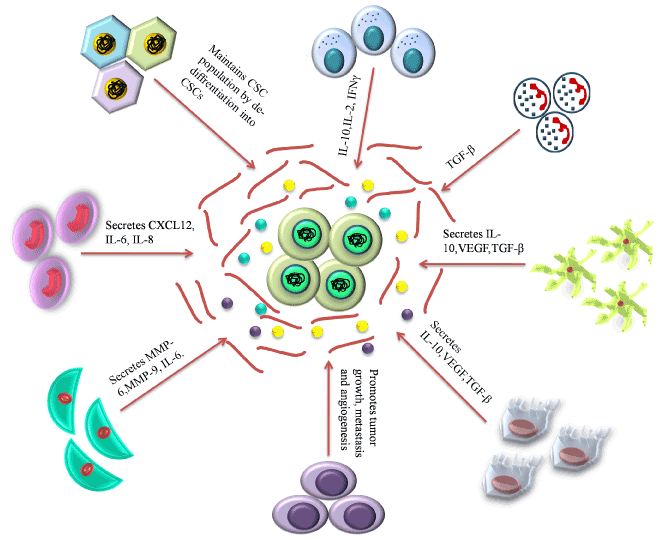
Figure 4. Role of niche in maintenance and proliferation of CSCs. Various cellular component of the CSC niche as well as the ECM play crucial roles in maintenance, growth and survival of the CSC population.
- Generated by hypoxia: Progressive tumor proliferation and the subsequent oxidative stress result in hypoxia within the tumor niche which in turn mediates the maintenance of stemness in the CSC population [120]. Hypoxia associated signalling molecules such as TGF-β, TNF-α and hypoxia inducible factor 1α and 2α (HIF1α, HIF2α) upregulate NOTCH and WNT signalling and downregulate c-Myc in order to retain stemness potential of the CSCs [83]. Wei et al. [121] demonstrated in 2011 that these hypoxic CSCs become capable of impeding the CD8+ T-cells within the tumor microenvironment.
- Shed by exosomes: Exosomes are membrane-derived vesicles of 30-200 nm diameters which carry proteins, RNAs, lipids and microRNAs to target cells and thus have critical roles in tumor invasion and metastasis [122,123]. The compositions of each exosome are closely linked to the functions of the cell from which they originate. These vesicles are formed through a complex process via the formation of multi-vesicular bodies (MVB) and then secreted through the plasma membrane via the process of exocytosis [124]. Drug resistant CSCs secrete exosomes to deliver P-gp and MRP1 transporters to the surrounding cancer cells as a process of reinforcing drug-resistance [83]. Several reports also highlighted the role of exosome in immuno-suppression [125] and also enhanced cytotoxic effect on immune cells [126] to make the environment more conducive for the CSCs. A study by Saghir et al. [127] showed that acute T cell leukemia (ATL)-derived exosomes delivered to MSCs results in the activation of NF-κB pathway along with its target genes which are involved in proliferation, angiogenesis and migration; this further modulates the MSCs to support ATL cells, by giving a more conducive environment for growth and metastasis.
As difficult as it is to develop specific targeted therapies against cancer due to the unshakable nature of the CSCs, researches have been done in the attempt to find alternative strategies. It is an intriguing idea to target the origin of the CSCs for therapeutic inventions. Our discussion highlights three possible origins of CSCs. Among those, targeting of non-stem cancer cells by numerous drugs-in-use has resulted in failure in abrogating CSCs. On the other hand, targeting normal stem cells may jeopardize the normal tissue homeostasis and regenerative capabilities of the body. In addition, since very little is known about VSELs, identifying processes to target them requires further research. At this point, the abstruse relationship between the tumor niche and the CSCs indicate that the niche can be targeted for therapeutic strategies in order to impede the survival of the CSCs.
TAFs have been trialled for anti-tumor therapeutic targets successfully. Inhibiting the polarization of TAFs by the recruitment of TNF-related apoptosis-inducing ligand (TRAIL) containing lipid-coated protamine DNA complexes (LPDs) resulted in apoptosis of the surrounding tumor cells followed by reprogramming of the TAFs [128]. DNA vaccines generated against the fibroblast activated protein also showed promise in MDR ct26 colon cancer model [129].
Many researchers have reported promising results by targeting the CXCL8-CXCR1/2 signalling of the TANs with antibodies [130]. A remarkable report in this context is by Acharyya et al. [131], which states increased sensitivity of both tumor and micro-metastases to chemotherapy after inhibition of CXCR2 in mouse metastatic breast cancer model. Verbeke et al. [132] showed that antibody-mediated inhibition of CXCL-6 interrupts TAN recruitment. Blocking the TGF-beta signalling pathway to promote anti-tumor TAN recruitment has also been an effective strategy [103]. Nevertheless, targeting neutrophils has potential risk of hampering the host defense system against infections. Therefore, it would be more feasible to target the reverse-migration mechanism of TANs by blocking the associated regulatory molecules like transinone-IIA [133]. Targeted TAN-specific nanoparticles and wound healing-associated macrophages that repel neutrophils, might also emerge as potential but safe tools for targeting TAN for anticancer treatment [134,135] (Figure 4).
Parallel to TAN-targeted anticancer strategies, targeting TAM recruitment with antibodies specific for CSF receptors has been attempted [136]. Depletion of the TAM and TAM progenitor population by bisphosphonates such as zoledronic acid and clodronate resulted in TAM apoptosis and the subsequent deterioration of malignancy in a mouse breast cancer model [137]. Another strategy for TAM targeting is reprogramming of TAMs to anti-tumor M1 phenotypes by the inhibition of the anti-inflammatory cytokine IL-10, or co-activating the pro-inflammatory CD40, IL-12, IL-8 and TNF-α by the antagonizing TLR-7 have also been effective in reverting tumor progression [138].
The Treg cells are also emerging as an intriguing subject for targeted cancer therapy. (cytotoxic T-lymphocyte associated protein 4) CTLA4 and (glucocorticoid induced TNFR family gene) GITR, constitutively expressed, Treg specific surface proteins, can be inhibited by humanized m-Ab and inhibitory drugs in order to enhance anti-tumor immune response from CD8+ and CD4+ T-cells [139-141]. But random depletion of the Treg cells poses a potential threat of autoimmune activation [142]. The effector Treg cells, marked by the overexpression of CCR4 (C-C motif chemokine receptor 4), are the predominant Tregs in the tumor niche [142]. Hence it is a better approach to particularly target the effector Treg cells by anti-CCR4 antibody in order to promote immune-activation within the tumor niche [143]. A report from our group has identified FOXP3 positive, IL10 positive T regulatory cells as the major pre-tumor Treg cells in the tumor microenvironment [144]. Depleting those Treg cells might be a good option of jeopardizing the CSC niche and therefore, CSC survival. In addition to the Treg depletion strategy, the anti-proliferative and DNA-damaging chemotherapeutic agents like cyclophosphamide also specifically affect the Treg cells when administered in low doses [145]. Several other reports present (T-cell receptor) TCR of the Treg cells as a potential target as it is differently regulated from that of the conservative T cells [146].
The ECM is also an exploitable subject for niche-dependent targeting of CSCs. Disruption of the interaction of the CSCs with the ECM by inhibiting the MMPs [147] and integrins [148] as well as by the application of collagenase and hyaluronidase have also shown promising results in hindering the disease progress [149].
Although the niche-targeted therapeutic strategies discussed above mostly reflect potential therapeutic efficacy on the tumor growth and development, these strategies indicate the possibility of indirectly targeting the CSCs due to their indispensable influence on CSC survival and maintenance.
It has been a baffling task to identify the essential root of cancer in order to develop an effective cure. Since the enthronement of the tumor-initiating CSCs, targeted therapeutic approaches against the CSCs have been employed in the hope of eradicating the disease as CSCs are reported to be the main factors behind disease initiation, metastasis, recurrence and therapeutic resistance. To target these highly resistant and aggressive stem-like cells, it becomes essential to identify their true origin. This review brings forth then on-stem cancer cells, the somatic stem cells and the VSELs as potential origins of the CSCs. However, identifying potential origin-specific therapeutic targets is a territory yet to be rigorously explored. On the other hand, the complex correlation between the CSCs and their corresponding niches also depicts the intriguing mystery of biological networking. There are cellular, environmental and soluble factors in the niche which protect the CSCs against chemotherapeutic and/or radioactive stresses. The recent discovery of exosomes secreted by the CSCs directly establishes their regulatory impact on the microenvironment. Therefore, this review points towards a possibility of niche-targeted therapy for killing highly resistant CSCs. In the end, the question remains as to the cancer stem cells or the niche - which is the ultimate amenable factor behind this refractory disease? Identification and subsequent confirmation of either of these two factors as the elixir of the disease can present us with the final target against which effective therapeutic strategies can be designed.
This work was supported by the research grants from Council of Scientific and Industrial Research (CSIR), University Grants Commission (UGC), Department of Biotechnology (DBT) and Department of Science and Technology (DST), Government of India.
- WHO (2017) Cancerfactsheets. http://www.who.int/mediacentre/factsheets/fs297.
- Hamilton G, Olszewski U (2013) Chemotherapy-induced Enrichment of Cancer Stem Cells in Lung Cancer. J Bioanal Biomed S9: 003.
- Lu H, Samanta D, Xiang L, Zhang H, Hu H, et al. (2015) Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc Natl AcadSci USA 112: e4600-e4609.
- Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M (2016) Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int 2016: 1740936. [Crossref]
- Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, et al. (2015) Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells 7: 1150-1184. [Crossref]
- Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS (2003) Stem cells in normal breast development and breast cancer. Cell Prolif 36 Suppl 1: 59-72. [Crossref]
- Zhang K, Guo Y, Wang X (2017) WNT/β-Catenin Directs Self-Renewal Symmetric Cell Division of hTERThigh Prostate Cancer Stem Cells. Cancer Res. [Crossref]
- Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 17: 165-172. [Crossref]
2021 Copyright OAT. All rights reserv
- Bradford GB, Williams B, Rossi R, Bertoncello I (1997) Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol 25: 445-453. [Crossref]
- Glauche I, Moore K, Thielecke L, Horn K, Loeffler M, et al. (2009) Stem cell proliferation and quiescence--two sides of the same coin. PLoS Comput Biol 5: e1000447. [Crossref]
- Goss PE, Chambers AF (2010) Does tumour dormancy offer a therapeutic target? Nat Rev Cancer 10: 871-877. [Crossref]
- Mohr M, Zänker KS, Dittmar T (2015) Cancer (stem) cell differentiation: An inherent or acquired property? Med Hypotheses 85: 1012-1018. [Crossref]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. 2003. Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821-5828.
- GursesCila HE, Acar M, Barut FB, Gunduz M, Grenman R, et al. (2016) Investigation of the expression of RIF1 gene on head and neck, pancreatic and brain cancer and cancer stem cells. Clin Invest Med 39: 27500. [Crossref]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl AcadSci USA 100: 3983-3988.
- Ejendal KF, Hrycyna CA (2002) Multidrug resistance and cancer: the role of the human ABC transporter ABCG2. Curr Protein Pept Sci 3: 503-511. [Crossref]
- Shaikh MV, Kala M, Nivsarkar M (2016) CD90 a potential cancer stem cell marker and a therapeutic target. Cancer Biomark 16: 301-307. [Crossref]
- Sreerama L, Sladek NE (1994) Identification of the class-3 aldehyde dehydrogenases present in human MCF-7/0 breast adenocarcinoma cells and normal human breast tissue. Biochem Pharmacol 48: 617-620. [Crossref]
- Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, et al. (2009) CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138: 286-299.
- Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, et al. (2007) CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A 104: 11008-11013. [Crossref]
- Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, Majeti R (2011) Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl AcadSci USA 108: 5009-5014.
- van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, et al. (2007) The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 110: 2659-2666.
- Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, et al. (2010) Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med 2: 17ra9. [Crossref]
- Hattermann K, Flüh C, Engel D, Mehdorn HM, Synowitz M, et al. (2016) Stem cell markers in glioma progression and recurrence. Int J Oncol 49: 1899-1910. [Crossref]
- Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, et al. (2008) Identification of cells initiating human melanomas. Nature 451: 345-349. [Crossref]
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, et al. (2014) SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511: 246-250. [Crossref]
- Li C, Liu S, Yan R, Han N, Wong KK, et al. (2017) CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics 7: 67-80. [Crossref]
- Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730-737. [Crossref]
- Haraguchi N, Ohkuma M, Sakashita H, Matsuzaki S, Tanaka F, et al. (2008) CD133+CD44+ population efficiently enriches colon cancer initiating cells. Ann Surg Oncol 15: 2927-2933. [Crossref]
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, et al. (2006) Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66: 7843-7848.
- Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, et al. (2006) Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res 66: 10247-10252. [Crossref]
- Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, et al. (2005) Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res 65: 5506-5511. [Crossref]
- Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442-454. [Crossref]
- Mukherjee S, Manna A, Bhattacharjee P, Mazumdar M, Saha S, et al. (2016) Non-migratory tumorigenic intrinsic cancer stem cells ensure breast cancer metastasis by generation of CXCR4(+) migrating cancer stem cells. Oncogene 35: 4937-4948. [Crossref]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, et al. (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438: 820-827. [Crossref]
- Ye X, Tam WL, Shibue T (2015) Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525: 256-260. [Crossref]
- Yang MH, Wu KJ (2008) TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle 7: 2090-2096. [Crossref]
- Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2: 48-58. [Crossref]
- Dean M (2009) ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 14: 3-9. [Crossref]
- Saha S, Mukherjee S, Khan P, Kajal K, Mazumdar M, et al. (2016) Aspirin Suppresses the Acquisition of Chemoresistance in Breast Cancer by Disrupting an NFκB-IL6 Signaling Axis Responsible for the Generation of Cancer Stem Cells. Cancer Res 76: 2000-2012. [Crossref]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, et al. (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756-760. [Crossref]
- Cheung TH, Rando TA (2013) Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 14: 329-340. [Crossref]
- Escoll M, Gargini R, Cuadrado A (2017) Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene. [Crossref]
- Yang M, Xiao X, Xing X, Li X, Xia T, et al. (2017) KRAS and VEGF gene 3'-UTR single nucleotide polymorphisms predicted susceptibility in colorectal cancer. PLoS One 12: e0174140. [Crossref]
- White AC, Lowry WE (2015) Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol 25: 11-20. [Crossref]
- Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, et al. (2013) Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev 27: 719-724. [Crossref]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, et al. (2014) Hippo pathway activity influences liver cell fate. Cell 157: 1324-1338. [Crossref]
- Iliopoulos D, Hirsch HA, Wang G, Struhl K (2011) Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A 108: 1397-1402. [Crossref]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, et al. (2007) Epigenetic stem cell signature in cancer. Nat Genet 39: 157-158. [Crossref]
- Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, et al. (2011) Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther 10: 325-335. [Crossref]
- Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, et al. (2010) Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6: 141-152. [Crossref]
- Yan GN1, Yang L, Lv YF, Shi Y, Shen LL, et al. (2014) Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol 234: 11-22. [Crossref]
- Jögi A, Øra I, Nilsson H, Lindeheim A, Makino Y, et al. (2002) Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl AcadSci U S A 99: 7021-7026.
- Xing F, Okuda H, Watabe M, Kobayashi A, Pai SK, et al. (2011) Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 30: 4075-4086. [Crossref]
- Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414: 105-111. [Crossref]
- Takebe N, Miele L, Harris PJ, Jeong W, Bando H, et al. (2015) Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 12: 445-464. [Crossref]
- Oh M, Nör JE (2015) The Perivascular Niche and Self-Renewal of Stem Cells. Front Physiol 6: 367. [Crossref]
- Pollina EA, Brunet A (2011) Epigenetic regulation of aging stem cells. Oncogene 30: 3105-3126. [Crossref]
- Bétous R, Renoud ML, Hoede C, Gonzalez I, Jones N, et al. (2016) Human Adipose-Derived Stem Cells Expanded Under Ambient Oxygen Concentration Accumulate Oxidative DNA Lesions and Experience Procarcinogenic DNA Replication Stress. Stem Cells Transl Med. [Crossref]
- Rodriguez R, Rubio R, Masip M, Catalina P, Nieto A, et al. (2009) Loss of p53 induces tumorigenesis in p21-deficient mesenchymal stem cells. Neoplasia 11: 397-407. [Crossref]
- Kucia M, Reca R, Campbell FR, Zuba-Surma E, Majka M, et al. (2006) A population of very small embryonic-like (VSEL) CXCR4(+)SSEA-1(+)Oct-4+ stem cells identified in adult bone marrow. Leukemia 20: 857-869. [Crossref]
- Kucia M, Halasa M, Wysoczynski M, Baskiewicz-Masiuk M, Moldenhawer S, et al. (2007) Morphological and molecular characterization of novel population of CXCR4+ SSEA-4+ Oct-4+ very small embryonic-like cells purified from human cord blood: preliminary report. Leukemia 21: 297-303. [Crossref]
- Zuba-Surma EK, Kucia M, Wu W, Klich I, Lillard JW Jr, et al. (2008) Very small embryonic-like stem cells are present in adult murine organs: ImageStream-based morphological analysis and distribution studies. Cytometry A 73A: 1116-1127. [Crossref]
- Chang YJ, Tien KE, Wen CH, Hsieh TB, Hwang SM (2014) Recovery of CD45(-)/Lin(-)/SSEA-4(+) very small embryonic-like stem cells by cord blood bank standard operating procedures. Cytotherapy 16: 560-565. [Crossref]
- Lee SJ, Park SH, Kim YI, Hwang S, Kwon PM, et al. (2014) Adult stem cells from the hyaluronic acid-rich node and duct system differentiate into neuronal cells and repair brain injury. Stem Cells Dev 23: 2831-2840. [Crossref]
- Shin DM, Zuba-Surma EK, Wu W, Ratajczak J, Wysoczynski M, et al. (2009) Novel epigenetic mechanisms that control pluripotency and quiescence of adult bone marrow-derived Oct4(+) very small embryonic-like stem cells. Leukemia 23: 2042-2051. [Crossref]
- Shin DM, Suszynska M, Mierzejewska K, Ratajczak J, Ratajczak MZ (2013) Very small embryonic-like stem-cell optimization of isolation protocols: an update of molecular signatures and a review of current in vivo applications. Exp Mol Med 45: e56. [Crossref]
- Ratajczak MZ, Ratajczak J, Suszynska M, Miller DM, Kucia M, et al. (2017) A Novel View of the Adult Stem Cell Compartment from the Perspective of a Quiescent Population of Very Small Embryonic-Like Stem Cells. Circ Res 120: 166-178. [Crossref]
- Kucia M, Zuba-Surma EK, Wysoczynski M, Wu W, Ratajczak J, et al. (2007) Adult marrow-derived very small embryonic-like stem cells and tissue engineering. Expert Opin Biol Ther 7: 1499-1514. [Crossref]
- Ratajczak MZ, Zuba-Surma EK, Machalinski B, Ratajczak J, Kucia M (2008) Very small embryonic-like (VSEL) stem cells: purification from adult organs, characterization, and biological significance. Stem Cell Rev 4: 89-99. [Crossref]
- Sovalat H, Scrofani M, Eidenschenk A, Pasquet S, Rimelen V, et al. (2011) Identification and isolation from either adult human bone marrow or G-CSF-mobilized peripheral blood of CD34(+)/CD133(+)/CXCR4(+)/ Lin(-)CD45(-) cells, featuring morphological, molecular, and phenotypic characteristics of very small embryonic-like (VSEL) stem cells. Exp Hematol 39: 495-505. [Crossref]
- Clevers H, Loh KM, Nusse R (2014) Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346: 1248012. [Crossref]
- Giovannini C, Bolondi L, Gramantieri L. 2016. Targeting Notch3 in Hepatocellular Carcinoma: Molecular Mechanisms and Therapeutic Perspectives. Int J Mol Sci 18: e56.
- Liu X, Ma M, Duan X, Zhang H, Yang M (2017) Knockdown of OCT4 may sensitize NSCLC cells to cisplatin. Clin Transl Oncol 19: 587-592. [Crossref]
- Garros-Regulez L, Garcia I, Carrasco-Garcia E, Lantero A, Aldaz P, et al. (2016) Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front Oncol 6: 222. [Crossref]
- Song C, Xu Q, Jiang K, Zhou G, Yu X, et al. (2015) Inhibition of BMK1 pathway suppresses cancer stem cells through BNIP3 and BNIP3L. Oncotarget 6: 33279-33289. [Crossref]
- Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, et al. (2010) Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther 10: 325-35.
- Kwiatkowska-Borowczyk EP, Gabka-Buszek A, Jankowski J, Mackiewicz A (2015) Immunotargeting of cancer stem cells. Contemp Oncol (Pozn) 19: A52-59. [Crossref]
- Schofield R (1978) The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4: 7-25. [Crossref]
- Li L, Xie T (2005) Stem cell niche: structure and function. Annu Rev Cell Dev Biol 21: 605-631. [Crossref]
- Morrison SJ, Spradling AC (2008) Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132: 598-611. [Crossref]
- Jones DL, Wagers AJ (2008) No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol 9: 11-21. [Crossref]
- Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16: 225-238. [Crossref]
- Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6: 392-401. [Crossref]
- Valenti G, Quinn HM, Heynen GJ, Lan L, Holland JD, et al. (2017) Cancer stem cells regulate cancer-associated fibroblasts viaactivation of Hedgehog signaling in mammary gland tumors. Cancer Res.
- Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, et al. (2013) Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer 110: 469-478.
- Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, et al. (1998) Tumor induction of VEGF promoter activity in stromal cells. Cell 94: 715-725. [Crossref]
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D (2010) Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 17: 135-47.
- Mao Y, Keller ET, Garfield DH, Shen K, Wang J (2013) Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev 32: 303-315. [Crossref]
- Meads MB, Gatenby RA, Dalton WS (2009) Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 9: 665-674. [Crossref]
- Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, et al. (2013) Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol 4: 400.
- Kharaziha P, Rodriguez P, Li Q, Rundqvist H, Björklund AC, et al. (2016) Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of Sorafenib against prostate cancer cells. Cell Death Dis 3: e262.
- Kitamura T, Qian BZ, Pollard JW (2015) Immune cell promotion of metastasis. Nat Rev Immunol 15: 73-86. [Crossref]
- Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, et al. (2015) Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci USA 112: e566-575.
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, et al. (2014) Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26: 638-652.
- Cabarcas SM, Mathews LA, Farrar WL (2011) The cancer stem cell niche--there goes the neighborhood? Int J Cancer 129: 2315-2327. [Crossref]
- Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496: 445-455. [Crossref]
- Noy R, Pollard JW (2014) Tumor-associated macrophages: from mechanisms to therapy. Immunity 41: 49-61. [Crossref]
- Smith HO, Stephens ND, Qualls CR, Fligelman T, Wang T, et al. (2013) The clinical significance of inflammatory cytokines in primary cell culture in endometrial carcinoma. Mol Oncol 7: 41-54. [Crossref]
- Su S, Liu Q, Chen J, Chen J, Chen F, et al. (2014) A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 25: 605-620. [Crossref]
- Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, et al. (2012) Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol 227: 17-28. [Crossref]
- Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A (2013) Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 228: 1404-1412. [Crossref]
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, et al. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell 16: 183-194. [Crossref]
- Raccosta L, Fontana R, Maggioni D, Lanterna C, Villablanca EJ, et al. (2013) The oxysterol-CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. Journal of Experimental Medicine 210: 1711-1728.
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, et al. (1999) Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nature Medicine 5: 828-831.
- Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, et al. (2012) Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology 56: 2242-2254.
- Toge H, Inagaki T, Kojimoto Y, Shinka T, Hara I (2009) Angiogenesis in renal cell carcinoma: the role of tumor-associated macrophages. Int J Urol 16: 801-807. [Crossref]
- Zhu Q, Wu X, Wang X (2017) Differential distribution of tumor-associated macrophages and Treg/Th17 cells in the progression of malignant and benign epithelial ovarian tumors. Oncology Letters 13: 159-166.
- Braga WM, Atanackovic D, Colleoni GW (2012) The role of regulatory T cells and TH17 cells in multiple myeloma. Clin Dev Immunol 2012: 293479. [Crossref]
- Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, et al. (2014) Neutrophils recruit regulatory T-cells into tumors viasecretion of CCL17—A new mechanism of impaired antitumor immunity. Int J Cancer 135: 1178-1186.
- Cuiffo BG, Karnoub AE (2012) Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell Adh Migr 6: 220-230. [Crossref]
- Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, et al. (2011) Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 71: 614-624. [Crossref]
- Reagan MR, Kaplan DL (2011) Concise review: Mesenchymal stem cell tumor-homing: detection methods in disease model systems. Stem Cells 29: 920-927. [Crossref]
- Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7: 733-736. [Crossref]
- Fessler E, Dijkgraaf FE, De Sousa E Melo F, Medema JP (2013) Cancer stem cell dynamics in tumor progression and metastasis: is the microenvironment to blame? Cancer Lett 341: 97-104. [Crossref]
- De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, et al. (2004) Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J 18: 1016-1018.
- Kessenbrock K, Dijkgraaf GJ, Lawson DA, Littlepage LE, Shahi P, et al. (2013) A role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell 13: 300-313. [Crossref]
- Hirata E (2016) Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments with High Integrin beta1/FAK Signaling. Cancer Cell 32: 127-137.
- Basak U, Dutta A, Dutta Chowdhury D, Bhattacharya A, Banerjee S, et al. (2017) Deciphering the Cancer Puzzle: Cancer Stem Cells Being the Pivotal Piece. J Stem cell Res Transplant 49: 1025
- Anido J, Sa ´ez-Borderi ´as A, Gonza `lez-Junca ` A, Rodo ´n L, Folch G, et al. (2010) TGF-b Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 18: 655-668.
- Wei J, Wu A, Kong LY, Wang Y, Fuller G, et al. (2011) Hypoxia potentiates glioma-mediated immunosuppression. PLoS One 6: e16195. [Crossref]
- Dutta S, Warshall C, Bandyopadhyay C, Dutta D, Chandran B (2014) Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS One 9: e97580. [Crossref]
- Yu S, Cao H, Shen B, Feng J (2015) Tumor-derived exosomes in cancer progression and treatment failure. Oncotarget 6: 37151-37168. [Crossref]
- Azmi AS, Bao B, Sarkar FH (2013) Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev 32: 623-642. [Crossref]
- Gutiérrez-Vázquez C, Villarroya-Beltri C, Mittelbrunn M, Sánchez-Madrid F (2013) Transfer of extracellular vesicles during immune cell-cell interactions. Immunol Rev 251: 125-142. [Crossref]
- Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M (2014) Sorting it out: regulation of exosome loading. Semin Cancer Biol 28: 3-13. [Crossref]
- El-Saghir J, Nassar F, Tawil N, El-Sabban M (2016) ATL-derived exosomes modulate mesenchymal stem cells: potential role in leukemia progression. Retrovirology 13: 73. [Crossref]
- Miao L, Liu Q, Lin CM, Luo C, Wang Y, et al. (2017) Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res 77(3):1-13.
- Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA (2006) Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. Journal of Clinical Investigation 116: 1955-1962.
- Khan MN, Wang B, Wei J, Zhang Y, Li Q, et al. (2015) CXCR1/2 antagonism with CXCL8/Interleukin-8 analogue CXCL8(3-72)K11R/G31P restricts lung cancer growth by inhibiting tumor cell proliferation and suppressing angiogenesis. Oncotarget 6: 21315-21327.
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, et al. (2012) A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150: 165-178. [Crossref]
- Verbeke H, Struyf S, Berghmans N, Van Coillie E, Opdenakker G, et al. (2011) Isotypic neutralizing antibodies against mouse GCP-2/CXCL6 inhibit melanoma growth and metastasis. Cancer Lett 302: 54-62. [Crossref]
- Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, et al. (2014) A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci Transl Med 6: 225ra29. [Crossref]
- Wang Z, Li J, Cho J, Malik AB (2014) Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotechnol 9: 204-210. [Crossref]
- Tauzin S, Starnes TW, Becker FB, Lam P, Huttenlocher A (2014) Redox and Src family kinase signaling control leukocyte wound attraction and neutrophil reverse migration. The Journal of Cell Biology 207: 589-598.
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, et al. (2014) Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25: 846-859. [Crossref]
- Rogers TL, Holen I (2011) Tumour macrophages as potential targets of bisphosphonates. J Transl Med 9: 177. [Crossref]
- Tang X, Mo C, Wang Y, Wei D, Xiao H (2013) Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 138: 93-104. [Crossref]
- Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, et al. (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364: 2517-2526. [Crossref]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, et al. (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. The Journal of Experimental Medicine 210: 1695-1710.
- Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S (2002) Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 3: 135-142. [Crossref]
- Tanaka A, Sakaguchi S (2017) Regulatory T cells in cancer immunotherapy. Cell Res 27: 109-118. [Crossref]
- Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, et al. (2013) Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proceedings of the National Academy of Sciences of the United States of America. 110: 17945-17950.
- Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, et al. (2013) FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity 39: 1057-69.
- Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, et al. (2007) Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer ImmunolImmunother 56: 641-648.
- Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, et al. (2012) T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37: 1-15.
- Tauro M, McGuire J, Lynch CC (2014) New approaches to selectively target cancer associated matrix metalloproteinase activity. Cancer Metastasis Rev 33: 1043-1057.
- Ruffini F, Graziani G, Levati L, Tentori L, D'Atri S, et al. (2015) Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of αvβ5 integrin. Int J Cancer 136: E545-558. [Crossref]
- Yata T, Lee EL, Suwan K, Syed N, et al. (2015) Modulation of extracellular matrix in cancer is associated with enhanced tumor cell targeting by bacteriophage vectors. Mol Cancer 14: 110. [Crossref]