Abstract
Introduction: Autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS) is a rare neurodegenerative disease characterized by cerebellar ataxia, peripheral neuropathy and pyramidal tract signs. Since its first report from Québec, more than 100 disease-causing variants have been reported in ARSACS, with variable clinical presentation. MRI imaging may help establishing the clinical diagnosis, especially if typical changes are present.
Clinical case: A 7-year old boy presented an early-onset, progressive ataxia, with sensory-motor neuropathy, nystagmus, feet deformities and cognitive impairment. MRI findings showed cerebellar and cervical spine atrophy and linear areas of hypointensity in the pons. The finding of a novel homozygous mutation - c.3066del(p.Asn1025Metfs*10) - in the SACS gene, resulting in a frameshift and a premature stop codon, allowed the genetic confirmation of the clinical diagnosis.
Key words
ataxia of Charlevoix-Saguenay, SACS, early-onset ataxia, spastic paraparesis, polyneuropathy
Introduction
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS, MIM 270550) is an early-onset neurodegenerative disease caused by mutations in SACS, a large gene on chromosome 13q12.12 [1,2]. This encodes a protein, sacsin, highly expressed in fibroblast, brain and skeletal muscle [3]. ARSACS originally found among inhabitants of the Charlevoix-Saguenay-Lac-Saint-Jean region of Québec [4], with an estimated incidence of 1/1932 habitants [5], but has since been increasingly reported worldwide [6,7]. The true global incidence of ARSACS is unknown, and it is probably underdiagnosed.
Classical clinical features include slowly progressive, early-onset (age 12-18 months), cerebellar ataxia,[8] sensory-motor neuropathy, pyramidal tract signs, finger and feet deformities, nystagmus and hypermyelination of retinal nerve fibres [9-12]. Cognition is usually preserved. Bladder and bowel symptoms have been commonly described, but not well-studied [4,10,12]. Recent reports on new phenotypic variants of ARSACS make the clinical diagnosis even more challenging [13,14]. There are currently no clinical diagnostic criteria for ARSACS.
Brain magnetic resonance imaging (MRI) may show atrophy of the superior cerebellar vermis, cervical spinal cord, corpus callosum and cortex, as well as areas of pontine hypointensity on T2-weighted section images [15]. Retinal nerve fibre hypertrophy, as demonstrated by ocular coherence tomography (OCT), has also been reported [16,17]. Nerve motor conduction velocities (NCMV) often confirms a mixed demyelinating and axonal neuropathy [4,18].
Two founder mutations, the c.8844del(p.Ile2949Phefs*4) single-base deletion (known as 6594del) and the c.7504C>T(p. Arg2502 *) nonsense mutation (known as 5254C>T), were identified in the individuals from Québec [19]. In a study of 164 alleles of patients with ARSACS born in Québec, Richter et al. (1999) reported that 92.6% cases were homozygous for the 6594 deletion [20]. Since the discovery of the gene responsible for ARSACS, more than 100 disease-causing genetic variants have been described around the world [7,10,12,21]. The authors present a case of a novel mutation related to ARSACS, in a Portuguese child.
Case report
A 7 year-old boy with a history of delayed walking (by age 32 months) complained of unsteady gait and frequent falls. Family history included consanguinity, but no history of neurological illness.
Clinical examination showed an ataxic gait, with appendicular dysmetria, and patellar and ankle hyperreflexia. Muscle tone and strength were preserved. Ocular pursuit examination showed mild gaze-evoked horizontal nystagmus, and no anomalies were seen at ophthalmoscopy. Cognitive assessment revealed a low IQ score (IQ of 60).
NMCV showed generalized axonal sensory neuropathy. Auditory evoked potentials showed bilateral prolongation of III and V waves with higher I-III and III-V intervals; visual evoked potentials were normal.
Brain and cervical T1-weighted MRI revealed cerebellar atrophy, especially in the upper vermis. T2-weighted and FLAIR axial section images showed linear pontine hypointensities (Figure 1). Optical coherence tomography showed no abnormalities.
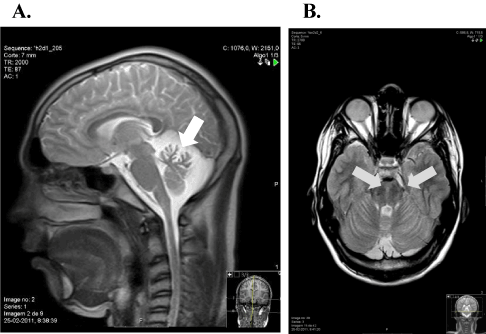
Figure 1. Brain and cervical spinal cord MRI images. A: sagittal T2-weight MRI image showing vermis cerebellar atrophy (white arrow). B: axial FLAIR MRI image at pons level with hypointense stripes in the paramedian pontine area (grey arrows)
The molecular study of the FXN gene GAA expansion and normal vitamin E values excluded Friedreich ataxia and AVED as possible diagnoses. A novel variant was found, in homozygosity, in exon 10 of the SACS gene, c.3066del(p.Asn1025Metfs*10), corresponding to a thymine deletion at position 3066, and leading to a frameshift resulting in a truncated protein (thus proving to be the cause of the disease). The parents, clinically normal, were heterozygous for that mutation.
The disease progressed with severe gaze-evoked horizontal nystagmus, with poor ocular pursuit, hyporeflexia of brachioradialis and ankle myotatic reflexes, extensor plantar reflexes, reduced pallesthesia and proprioception, and feet deformities (claw fingers and ulcer lesions) (Figure 2a). Treatment with a combination of baclofen (30mg daily) and botulinum toxin was established at age 15 years. In 19 years, he presented with gait with steppage, needing orthotics support, and bladder dysfunction (Figure 2b). Cardiovascular and ophthalmological exam remained normal at that stage.
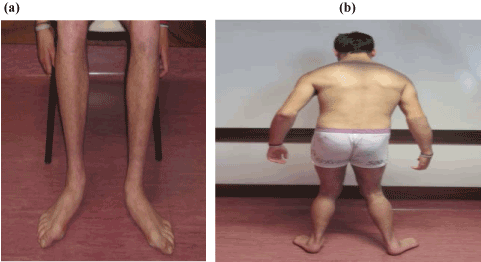
Figure 2. (a) Severe amyotrophy and feet deformities (claw fingers); (b) orthostatism is achieved through a broad base support posture with an anterior flexure of the trunk. Hip and knee muscle retraction prevents full leg extension.
Discussion
ARSACS is a complex recessive and progressive ataxia. Age of onset and clinical presentation in this case were similar to the initial reports, except for cognitive disability, described only occasionally in reports outside Québec [22,23]. IQ score, when normal, tend to be in the lower range. Most affected individuals are able to manage daily tasks [5].
Retinal changes were a2021 Copyright OAT. All rights reservns of ARSACS outside Québec also reported normal retinal fibres [7,10]. Neuropathy usually presents after the 2nd decade of life and progresses to spastic paraparesis;[9] in that case, the authors highlight the early onset of neuropathy, with rapidly progressive distal weakness, resulting in loss autonomous gait.
Typical MRI changes helped establishing the diagnosis in this case [15,24]. Electrophysiological findings, such as the demyelinating component of the sensorimotor peripheral neuropathy, may help distinguishing ARSACS from common recessive ataxias like Friedreich ataxia.
Predicting the clinical implications of novel variants can be challenging and variants of uncertain clinical significance (VUS) are frequent in SACS [10]. This c.3066del(p.Asn1025Metfs*10) genetic variant is not present in population databases and is predicted to produce a truncated protein; it is, thus, most probably disease-causing and likely to be associated with this more aggressive early-onset phenotype, with loss of autonomous gait by the 2nd decade. Follow-up of this patient will contribute to elucidate this.
ARSACS should be evoked in the differential diagnosis of children presenting with early onset ataxia and spastic paraparesis. Supportive treatment is available for ARSACS patients. Genetic counselling and carrier testing must be proposed to adult relatives of these patients.
References
- Bouchard JP, Richter A, Mathieu J, Brunet D, Hudson TJ, et al. (1998) Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuromuscul Disord 8: 474-479. [Crossref]
- Engert JC, Doré C, Mercier J, Ge B, Bétard C, et al. (1999) Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): high-resolution physical and transcript map of the candidate region in chromosome region 13q11. Genomics 62: 156-164. [Crossref]
- Engert JC, Bérubé P, Mercier J, Doré C, Lepage P, et al. (2000) ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 24: 120-125. [Crossref]
- Bouchard JP, Barbeau A, Bouchard R, Bouchard RW (1978) Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 5: 61-69. [Crossref]
- Vermeer S, van de Warrenburg BP, Kamsteeg EJ (2003) ARSACS. 2003 [Updated 2012 Oct 11]. In: Pagon RA, Adam MP, Ardinger HH. GeneReviews®. [http://www.ncbi.nlm.nih.gov/books/NBK1255/].
- Gomez CM (2004) ARSACS goes global. Neurology 62: 10-11. [Crossref]
- Baets J, Deconinck T, Smets K, Goossens D, Van den Bergh P, et al. (2010) Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 75: 1181-1188. [Crossref]
- Bouchard JP (1991) Recessive spastic ataxia of Charlevoix-Saguenay. In: de Jong JMBV edn. Handbook of Clinical Neurology 16: Hereditary Neuropathies and Spinocerebellar Degenerations 60. Amsterdam, Netherlands: Elsevier: 451-459.
- Duquette A, Brais B, Bouchard JP, Mathieu J (2013) Clinical presentation and early evolution of spastic ataxia of Charlevoix-Saguenay. Mov Disord 28: 2011-2014. [Crossref]
- Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, et al. (2013) Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 8: 41. [Crossref]
- Anheim M, Tranchant C, Koenig M (2012) The autosomal recessive cerebellar ataxias. N Engl J Med 366: 636-646. [Crossref]
- Prodi E, Grisoli M, Panzeri M, et al. (2013) Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur J Neurol 20: 138-146. [Crossref]
- Bouhlal Y, Amouri R, El Euch-Fayeche G, Hentati F (2011) Autosomal recessive spastic ataxia of Charlevoix-Saguenay: an overview. Parkinsonism Relat Disord 17: 418-422. [Crossref]
- Takiyama Y (2007) Sacsinopathies: sacsin-related ataxia. Cerebellum 6: 353-359. [Crossref]
- Martin MH, Bouchard JP, Sylvain M, St-Onge O, Truchon S (2007) Autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of MR imaging in 5 patients. AJNR Am J Neuroradiol 28: 1606-1608. [Crossref]
- Vingolo EM, Di Fabio R, Salvatore S, Grieco G, Bertini E, et al. (2011) Myelinated retinal fibers in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Eur J Neurol 18: 1187-1190. [Crossref]
- Pablo LE, Garcia-Martin E, Gazulla J, Larrosa JM, Ferreras A, et al. (2011) Retinal nerve fiber hypertrophy in ataxia of Charlevoix-Saguenay patients. Mol Vis 17: 1871-1876. [Crossref]
- García A, Criscuolo C, de Michele G, Berciano J (2008) Neurophysiological study in a Spanish family with recessive spastic ataxia of Charlevoix-Saguenay. Muscle Nerve 37: 107-110. [Crossref]
- Mercier J, Prevost C, Engert JC, Bouchard JP, Mathieu J, et al. (2001) Rapid detection of the sacsin mutations causing autosomal recessive spastic ataxia of Charlevoix-Saguenay. Genet Test 5: 255-259. [Crossref]
- Richter A, Rioux JD, Bouchard JP, Mercier J, Mathieu J, et al. (1999) Location score and haplotype analyses of the locus for autosomal recessive spastic ataxia of Charlevoix-Saguenay, in chromosome region 13q11. Am J Hum Genet 64: 768-775. [Crossref]
- Gazulla J, Benavente I, Vela AC, Marín MA, Pablo LE, et al. (2012) New findings in the ataxia of Charlevoix-Saguenay. J Neurol 259: 869-878. [Crossref]
- Verhoeven WM, Egger JI, Ahmed AI, Kremer BP, Vermeer S, et al. (2012) Cerebellar cognitive affective syndrome and autosomal recessive spastic ataxia of charlevoix-saguenay: a report of two male sibs. Psychopathology 45: 193-199. [Crossref]
- Takiyama Y (2006) Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuropathology 26: 368-375. [Crossref]
- Gerwig M, Krüger S, Kreuz FR, Kreis S, Gizewski ER, et al. (2010) Characteristic MRI and funduscopic findings help diagnose ARSACS outside Quebec. Neurology 75: 2133. [Crossref]