We report a case of a 41-year-old woman with refractory high blood pressure sustained by an unexpected combination of a pheochromocytoma (PCC) and reflux nephropathy. This case prompted us to seek a molecular explanation for this unusual association of VUR and a PCC, as they are derived from distinct embryonic tissues. Both entities can be caused by over expression of the RET gene, and this might be the cause of this association.
high blood pressure, pheochromocytoma, RET gene, vesicoureteral reflux
Refractory high blood pressure (HBP) is a serious disease associated with a particularly high cardiovascular risk. Its prevalence is about 20% among all hypertensive patients [1]. An etiology of HBP is found in only 10-20% of cases. Etiologies are more often found when the HBP is refractory [1].
We report the case of a patient with refractory HBP sustained by an unexpected combination of a pheochromocytoma (PCC) and reflux nephropathy.
A 41-year-old woman was admitted to our department for the management of refractory HBP. She had had the disease for the previous 7 years, and had been treated with many antihypertensive agents. At the time of her admission, she was being treated with 3 drugs: Amlodipine (10 mg per day), Indapamide (2.5 mg per day) and Losartan (100 mg per day). The previous minimal investigation was normal. Diabetes mellitus was diagnosed six months before hospitalization.
Her family history did not reveal anything significant. A physical examination revealed an afebrile patient, with a body mass index of 20.93 kg/m2, blood pressure 180/120 mmHg in both arms and a heart rate of 92 beats/min. A cardiovascular examination revealed a normal cardiac auscultation and all pulses were present with symmetric breathlessness on vascular routes. Diuresis was normal and the dipstick was negative. Laboratory findings were normal, with negative proteinuria and hematuria. Serum creatinine was at 65 µmol/l (MDRD 93 ml/mn/1,73m2). The electrocardiogram and heart sounds did not reveal left ventricular hypertrophy. However a stage 2 hypertensive retinopathy was noticed.
An abdominal ultrasound revealed a small dedifferentiated left kidney of 40mm x 20mm and a normal right kidney of 120mm x 60mm with good cortico-sinusal differentiation. No adrenal mass was noticed. A renal artery doppler was conducted and excluded stenosis of the left renal artery. A subsequent retrograde urethrocystography showed a left paraureteral diverticulum with an active left vesico-ureteral reflux (VUR) grade 3 (Figure 1).
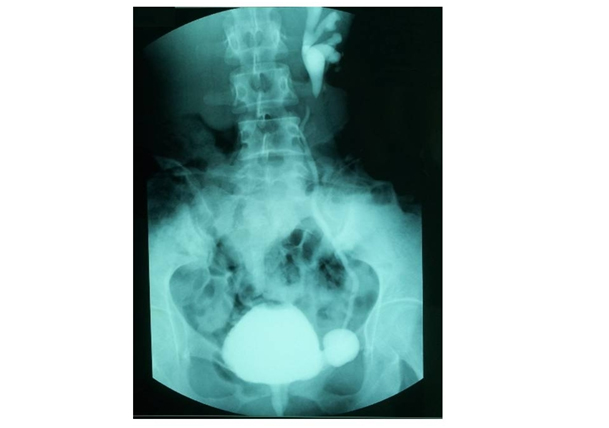
Figure 1. Retrograde urethrocystography
Active left vesico- ureteral reflux grade 3 and left paraureteral diverticulum
It was evident from the ultrasound that the left kidney was totally destroyed and almost certainly non-functional; it was assumed to be responsible for the refractory hypertension, in addition to renal parenchymal disease due to VUR. These conditions led to surgical kidney removal.
A 5 cm adrenal mass, causing on touch a hypertensive peak, was discovered intraoperatively. It was decided to perform a left adrenalectomy during the same operation. Histopathologic examination of the resected kidney showed marked tubular atrophy with secondary sclerosis of the glomeruli (Figure 2A). Adrenal mass examination revealed a highly vascularized tumor proliferation made of layers of enlarged cells provided with abundant cytoplasm (Figure 2B). The immunohistochemical study showed positivity of tumor cells with antibodies anti-chromogranin, anti-synaptophysin and anti-CD56, confirming the diagnosis of a pheochromocytoma.
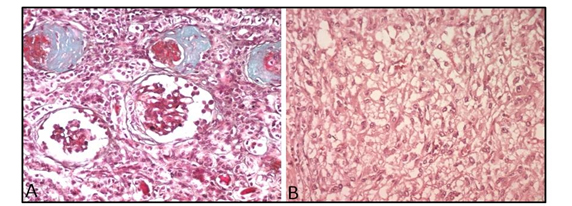
Figure 2. Histopathological examinations
A) Histopathological examination of the resected kidney: marked tubular atrophy with secondary sclerosis of the glomeruli (Trichrome, x200) B) Histopathological examination of the adrenal mass: highly vascularized tumor proliferation made of layers of enlarged cells provided with abundant cytoplasm (H&E, x200).
From the day after the operation until her recent 10 month post-operative follow-up, the patient had normal blood pressure and was not prescribed any anti-hypertensive medication.
A PCC is a tumor developed by chromaffin cells of the adrenal medulla or other sympathetic ganglia and varied secreted amounts of catecholamines. A PCC needs to be considered in the case of symptomatic paroxysmal hypertension. It may also be associated with orthostatic hypotension and diabetes.
Our patient had none of these clinical signs. Indeed a PCC may be asymptomatic, suggesting a desensitization of the cardiovascular system to circulating catecholamines and may thus be detected incidentally by imagery [2]. As in the case of our patient, a PCC can be discovered intraoperatively following a massive release of catecholamines [3]. Currently, 50% of PCC cases are diagnosed during a postmortem [4] thereby confirming that asymptomatic PCCs are not uncommon.
Reflux nephropathy is often, but not always, caused by intrarenal reflux of infected urine. In turn, it triggers varied blood pressure, immunological phenomena and inflammation. This results in a focal and segmental ischemic glomerulosclerosis associated with interstitial fibrosis with atrophy. While reflux nephropathy is responsible for renal parenchymal HBP in 40% of cases, a pheochromocytoma (PCC) is the cause of only 0.1 to 6% of HBP cases [5].
VUR in our patient was congenital as revealed by the presence of a bladder diverticulum. To our knowledge, the association of a PCC with VUR has been previously described only once [6], however only the abstract of the article is available. Saito et al. assumed, in that abstract, the existence of a high plasma nor-epinephrine concentration was implicated in the pathogenesis of segmental and focal glomerular sclerosis lesions noticed during reflux [6]. This case prompted us to seek a molecular explanation for this unusual combination. Prima facie, VUR and PCC seem to be sustained by distinct developmental pathways: PCC originates from neural crest cells, hence from the ectoblast, while the urinary tract originates from the mesonephrotic ducts.
At the genetic level, PCC is associated with hereditary syndromes such as multiple endocrine neoplasia type 2 caused by mutations in the RET gene, von Hippel-Lindau disease associated with germline mutation of the VHL gene, neurofibromatosis type 1 syndrome with involvement of the NF1 gene, and pheochromocytoma/paraganglioma syndrome strongly related to the SDHB and SDHD genes. Recent examinations have demonstrated the involvement of newly discovered genes i.e. KIF1Bβ, MAX and TMEM127 in the pathogenesis of PCC [7].
Our patient had no family history of PCC and no clinical signs of the syndromes cited above. One approach to understand how VUR and PCC could occur simultaneously is to examine genes involved in the development of both entities. In reviewing shared molecular pathways between PCC and VUR, RET gene activation seems to be a plausible hypothesis to explain this simultaneous occurrence [8].
The RET gene is significant for urinary tract development [9,10]. When over expressed, ureteral insertion into the bladder is affected, so the flap-valve mechanism fails to mature leading to postnatal VUR.RET mutations were also assumed to be responsible for renal agenesis and dysgenesis [11,12]. Moreover the RET gene is known to play an essential role in the development of the parasympathetic as well as sympathetic nervous systems. PCC, a part of the sympathetic system, is associated with RET gene over expression [7]. As reported by Griseri et al., RET expression was strongly decreased in a patient with a complex clinical presentation combining Hirschsprung disease (HD) and vesico-ureteral reflux [13].
As with PCC, HD is sustained by the intestinal aganglionosis part of the sympathetic nervous system, strongly implying the RET gene. Thus neural crest derived tumors would be linked to RET over expression, while absence of neural crest cells illustrated by aganglionosis in the case of HD would be linked to decreased expression of RET.
This case indicates that PCC and VUR can occur simultaneously; both entities can be caused by over expression of the RET gene, and this might be the cause of this association. Molecular investigations targeting the RET gene are being carried out on our patient to further investigate this hypothesis
When hypertension persists even after PCC removal, we should consider the possibility of VUR. Conversely, patients with refractory hypertension and VUR should be examined for the presence of PCC.
The authors are very grateful to the patient who kindly participated in this study.
The authors declare that they have no conflicting interests and no financial disclosures. The results presented in this paper have not been published previously in whole or part, except in abstract format.
- Wilson PW (1994) Established risk factors and coronary artery disease: the Framingham Study. Am J Hypertens 7: 7S-2S. [Crossref]
- Prys-Roberts C (2000) Phaeochromocytoma--recent progress in its management. Br J Anaesth 85: 44-57. [Crossref]
- Steppan J, Shields J, Lebron R (20) Pheochromocytoma presenting as acute heart failure leading to cardiogenic shock and multiorgan failure. Case Rep Med 20: 596354. [Crossref]
- Plouin PF, Gimenez-Roqueplo AP, La Batide Alanore A, Salenave S, Duclos JM (2000) Recent progress in the diagnosis, prognostic evaluation and treatment of pheochromocytomas. Rev Med Interne 2: 075-085. [Crossref]
- Bravo EL (994) Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev 5: 356-368. [Crossref]
- In H, Saito T, Nishi S, Maruyama Y, Takayama R, et al. (99) A case of reflux nephropathy associated with pheochromocytoma. Nihon Jinzo Gakkai Shi 33: 35-40. [Crossref]
- KolaÄkov K, Tupikowski K, Bednarek-Tupikowska G (202) Genetic aspects of pheochromocytoma. Adv Clin Exp Med 2: 82-829. [Crossref]
- Yu OH, Murawski IJ, Myburgh DB, Gupta IR (2004) Overexpression of RET leads to vesicoureteric reflux in mice. Am J Physiol Renal Physiol 287: F23-F30. [Crossref]
- Van Eerde AM, Duran K, Van Rie E, de Kovel CG, Koeleman Bp, et al. (2012) Genes in the Ureteric Budding Pathway: Association Study on Vesico-Ureteral Reflux Patients. Plos one 7: e3327. [Crossref]
- Hibi Y, Ohye T, Ogawa K, Shimizu Y, Shibata M, et al. (204) A MEN2A family with two asymptomatic carriers affected by unilateral renal agenesis. Endocr J 6: 9-23. [Crossref]
- Yin L, Puliti A, Bonora E, Evangelisti C, Conti V, et al. (2007) C620R mutation of the murine ret proto-oncogene: loss of function effect in homozygotes and possible gain of function effect in heterozygotes. Int J Cancer 2: 292-300. [Crossref]
- Sweetser DA, Froelick GJ, Matsumoto AM, Kafer KE, Marck B, et al. (999) Ganglioneuromas and renal anomalies are induced by activated RET(MEN2B) in transgenic mice. Oncogene 8: 877-886. [Crossref]
- Griseri P, Vos Y, Giorda R, Gimelli S, Beri S, et al. (2009) Complex pathogenesis of Hirschsprung's disease in a patient with hydrocephalus, vesico-ureteral reflux and a balanced translocation t(3;7)(p2;q). Eur J Hum Genet 7: 483-490. [Crossref]