This paper proposes a simple and selective UHPLC method for determination of degradation products of a model pyrrole based compound, containing susceptible to hydrolysis ester group. The chromatographic separation was achieved on BDSHYPERSILC18 (150 mm х 4.6 mm, 3.5 µm) column thermostated at 30°C under gradient elution by a binary mixture of potassium dihydrogen phosphate (pH 3.0, 0.02 M) and acetonitril at a flow rate of 0.8 ml/min. A UV/Vis detector set at 225 nm was used for detection. The stability testing was carried out under acidic (pH 1.2 and pH 4.5), normal (pH 6.8) and alkaline (pH 13) conditions at temperature of 37°C. At moderate and low alkali pH and temperatures the compound was stable. In acidic media the model compound is transformed into its degradation product. The developed method is validated with respect to suitability, linearity, precision, accuracy and selectivity and can be implemented for stability testing of pyrrole-containing ester derivatives.
Chemical Stability, Hydrolysis, Physiological Stability, Pyrrole
The stability of a compound synthesized as a potential medicinal agent is related to the pharmacokinetic behavior in the body and to the conditions for the formulation, storage, occurrence of toxic effects associated with degradation products and so on. Most of compounds are fairly stable in the neutral pH value found in the intestine but can be unstable at the pH value found in the stomach [1]. Thus in the development of new drugs early information of stability becomes essential for subsequent processes of optimization and selection of leading active structures and can prevent unnecessary costs on developing products that subsequently prove to be unstable.
In this regard, the synthesis and determination of stability at different physiological conditions of the new compounds with potential anti-inflammatory activity is of particular importance. Forced degradation testing performed according to the regulatory guidance suggests, that potential degradation products are formed through elevated temperatures, by applying acidic, basic, and oxidative conditions, and through photolysis [2]. Several high-performance liquid chromatography (HPLC) methods are reported in the literature for the identification and quantification of degradation of different pharmaceutical substances in various media [3-8].
The aim of this study is to establish and apply a ultra-high performance liquid chromatography (UHPLC) method for identification of hydrolytic degradation products from newly synthesized pyrrolyl-ester derivative.
Chemicals and Reagents
All commercial chemicals used in this study as starting materials and reagents were purchased from “Merck” (Darmstadt, Germany). The melting point was determined with a capillary digital melting point apparatus IA 9200 Electrothermal AZ9003MK4, South end-on-Sea, UK. The IR spectra were registered on Specord IR-71, Carl Zeiss, Jena, Germany (KBr). The 1Н NMR spectra (250 MHz, 20oC) was registered on a BrukerSpectrospin WM250 spectrometer (Faenlanden, Switzerland), using TMS as internal standard. TLC characteristic of the product was measured on aluminum sheets of silica gel 60 F254, Merck 1.05554 at ambient temperature using a mobile phase chloroform-ethanol.
The necessary products for preparation of the mobile phase and used buffers are of analytical grade, whereas potassium dihydrogen phosphate dihydrate (Sigma-Aldrich, Steinheim, Germany), orthophosphoric acid (Merck, Darmstadt, Germany) and acetonitrile (ACN) gradient grade (Sigma-Aldrich, Steinheim, Germany) were used.
Instrumentation and chromatographic conditions
The chromatographic analysis was performed on a system UHPLCDionexUltiMate 3000 with UV/Vis detector (Shimadzu, Kyoto, Japan). A BDSHYPERSILC18 (150 mm х 4.6 mm, 3.5 µm) column was employed for separation and isolation of degradation product using acetonitrile (CH3CN)/phosphate buffer (pH 3.0, 0.02 M) 40/60 (v/v) as mobile phase A and CH3CN as mobile phase B with a gradient elution with flow rate of 0.8 ml/min and detection at 225 nm (Table 1). The column oven was conditioned at 30°C. The injection volume was 20 μl with analysis time of 25 minutes.
Table 1. Linear gradient program.
Time [min] |
Flow [ml/min] |
% A |
% B |
0.0 |
0.8 |
100 |
0 |
5.0 |
0.8 |
100 |
0 |
12.5 |
0.8 |
25 |
75 |
20.0 |
0.8 |
25 |
75 |
22.0 |
0.8 |
100 |
0 |
25.0 |
0.8 |
100 |
0 |
Preparation of buffer component of mobile phase
2.70 g of potassium dihydrogen phosphate dihydrate was dissolved in 1μL of ultrapure water. An orthophosphoric acid solution (6%) was used to adjust the pH to 3.0 (±0.05). The mobile phase buffer was filtered through a membrane filter (0.20 μm) using a Millipore glass filter holder. The mobile phase buffer was used immediately after preparation or stored in the refrigerator in closed borosilicate glass bottles for a maximum of 24 hours
Mobile phase composition:
Mobile phase A - CH3CN/phosphate buffer pH 3.0 = 40/60 (v/v).
Mobile phase B - CH3CN.
The aim of this study is to identify, validate and apply a gradient UHPLC method for determination of chemical and physiological stability of pyrrole-containing ester derivative.
Selection of a model compound
The fully substituted pyrrole, as well as the connected with it methyl groups are stable at moderate temperatures and in wide range of pH values. In previous studies, we have synthesized many pyrrole-based compounds bearing a variety of functional groups, like ethoxy-carbonyle, acetyl, carboxamide etc on 3rd position in the heterocycle [9]. Thus we considered that the stability of the obtained structures will be determined by the stability of the identified as potential “vulnerable” groups susceptible to hydrolysis ester and to lower extend amide groups positioned on 3rd place in the pyrrole cycle (Figure 1)[10].
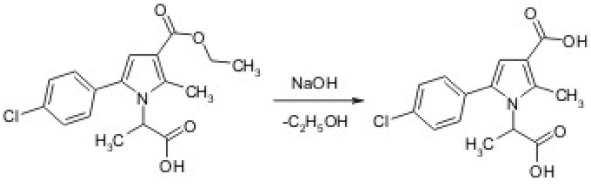
Scheme 1 A synthetic access to the targeted product 2A.
In order to confirm this 1A was chosen as a model compound [11]. The structure of the analyzed molecule is presented on Figure 1.
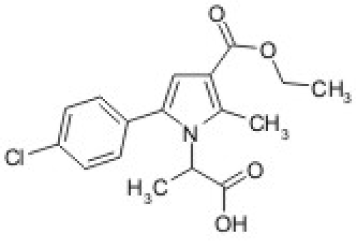
Figure 1. Chemical structure of the chosen model compound 1A.
Synthesis of the hydrolysis product
The hydrolysis of the ester substituent at 3rd position into the corresponding carboxylic group was defined as the most probable chemical transition under the evaluated conditions.
Thus as referent compound the corresponding di-carboxylic acid 2A, where the ester group on 3rd position is replaced with the carboxylic group was synthesized. The structures of the evaluated ester and its hydrolitical acid product are presented on Figure 2.
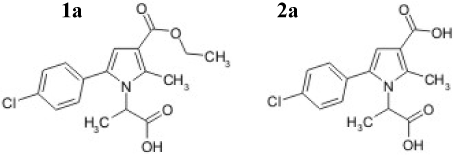
Figure 2. Structure of the evaluated compound (1A) and its probable hydrolytic product (2A).
The reference product was obtained by alkaline hydrolysis according to the described synthetic procedure where 0.10 moles of 1-(1-Carboxy-ethyl)-5-(4-chloro-phenyl)-2-methyl-1H-pyrrole-3-carboxylic acid ethyl ester (1A) were dissolved in ethanol (140 ml) and 25% NaOH (70 ml) were added. The reaction was run at the water bath boiling point and the process was monitored by thin layer chromatography (TLC). After reaction completion (2 hours), the mixture was cooled down and 20 % HCl were added, until pH 2.0. The separated precipitate was filtered off, washed with water to pH 7.0, dried and recrystallized from warm ethanol. The structure of the obtained product was elucidated by the corresponding IR and 1H-NMR spectral data. The yield was calculated for the chromatographically pure product (Scheme 1) [12].
1-(1-Carboxy-ethyl)-5-(4-chloro-phenyl)-2-methyl-1H-pyrrole-3-carboxylicacid 2A): White solid, Yield 70%, mp 194-196оС, Rf0.63 (10:0.3/CHCl3:C2H5OH). IR spectrum (KBr), n, cm-1: 3600-2400 (COOH), 3300 (O-H), 1720, 1695 (C=O), 830 (p-C6H4); 1H NMR spectrum (250 MHz,DMSO-d6),d, ppm (J, Hz):1.55 (d,3H, J=7.1, CHCH3); 2.50 (s,3H, CH3-2), 4.60-4.80 (m,1H, CHCOOH), 6.40 (s,1H, H-4), 7.15-7.25 (m,4H,C6H4), 11.90 ppm – bs, 2H, OH (OH protons were D2O exchangeable).
Validation of the gradient UHPLC analytical procedure
The method was validated according to ICH Q2 (R1) guidelines [13]. The system suitability (i.e., repeatability of retention times and areas, number of theoretical plates, and resolution), precision, linearity, accuracy and selectivity were evaluated during method validation (Table 2). The parameters accuracy, precision, and selectivity were evaluated for the two analyzed products.
Table 2. Validation parameters for compounds 1A and 2A.
|
1A |
2A |
Criterion |
Repeatability tR (% RSD)* |
0.00 |
0.00 |
X < 1% |
Number of Theoretical plates |
50 000 |
10 000 |
- |
Resolution* |
7.3 |
7.3 |
Rij > 1.5 |
Precision (% RSD)# |
1.0 |
1.0 |
X < 5% |
Linearity (correlation coefficient)§ |
0.9997 |
0.9997 |
R > 0.9990 |
Accuracy (%)# |
100.1 |
99.97 |
X = 100 ± 5% |
Selectivity |
No interference |
No interference |
No interference |
* Six injections.
# Two samples, three injections of each sample.
§ At 50, 100, 150, 200 and 250 % concentration level.
% RSD: Relative standard deviation in %
Precision: Six sample solutions were prepared from each of the evaluated substances. Each sample was injected three times. The final results are reported as relative standard deviations (RSD) of the 1A and 2A ratios of the peak areas.
Linearity: A 5 points calibration curve was create, covereing the concentration range of 1A from 0.02 mg/ml to 0.1 mg/ml. Linear regression was used to process the calibration data. The correlation coefficients of linearity was 0.9997 for 1A, which indicate good correlation between the peak areas and the range of concentrations studied.
Accuracy: The solutions for injection were prepared using a placebo and stock solution of the tested structures. Six solutions were prepared from each of the two compounds. Each solution was injected onto the column three times. Accuracy is reported as a parameter recovery with relative standard deviations.
Selectivity: The selectivity was determined by comparing the chromatograms of sample solutions, standard solution, and blank solutions. Figure 3 shows that the two tested compounds are all completely separated from each other. No interference was observed.
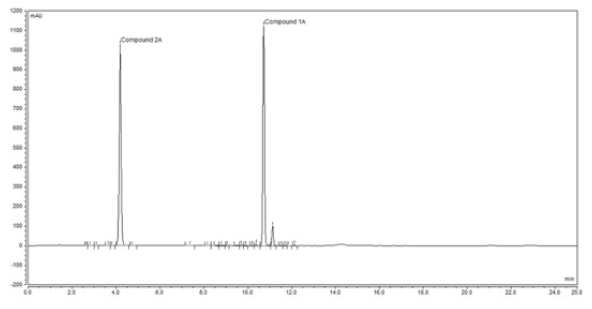
Figure 3. Retention times of the hydrolytic product 2А (4.2 min.) and the evaluated compound 1А (11.0 min.), respectively.
Determination of the stability at different рН
Chemical stability: By definition the chemical stability is determined as the tendency of a substance to sustain to changes or decay, caused by internal reaction or affected by air, humidity, heat, light, pressure etc. The compound presented in this paper has been stored for 6 months period at room temperature with access to air and light. The physical and chemical properties of the compound were determined to be unchanged under these conditions. The tested compound may be considered chemically stable.
Physiological stability: An important factor influencing the performance of the molecules in the organism is their hydrolytic stability at physiological conditions, such as: body temperature of 37oC and physiological pH of 1.2 (in stomach), 4.5 (in urine) and 6.8 (in blood plasma) [14].
Hydrolytic stability study at close to physiological conditions
Due to the poor solubility of the analyzed structure in water, for the evaluation of the stability anacetonitrile:buffer solutions were prepared at the necessary relevant ratio, in order to obtain the desired pH values, close to physiological. A 20 mg sample of the model compound was weighed and dissolved in the corresponding mixture of acetonitrile: buffer 1.2, 4.5, 6.8 and 13, respectively. The obtained solutions were termostated and stirred in a micro reactor at 37oC for a total time of 1440 min. An aliquot samples of 0.5 ml of the analyzed solutions were drawn at definite time intervals (15, 30, 60, 120, 240, 480 and 1440 min) and diluted up to 1.5 ml with the corresponding mobile phase in a way that the concentration is in the range of 0.1 mg/ml and a 20 μl sample was injected. The corresponding chromatograms were obtained.
Stability determination of compound 1А at рН 1.2 (stomach) and temperature of 37ºС
Preparation of buffer pH 1.2: A mixture of 0.2 mol/l NaCl (solution A) and 0.2 mol/l HCl (solution B) is prepared, where 25 ml of solution A are mixed with 42.5 ml of solution B. The obtained mixture is diluted up to 100 ml with distilled water.
A 20 mg sample of the model compound was weighed and dissolved in the corresponding mixture of acetonitrile: buffer pH 1.2. The obtained solutions were stirred in a micro reactor at 37oC for a total time of 1440 min. An aliquot 20 μl samples were drawn at the defined above time intervals and injected into the UHPLC system. The obtained chromatogram is presented on Figure 4.
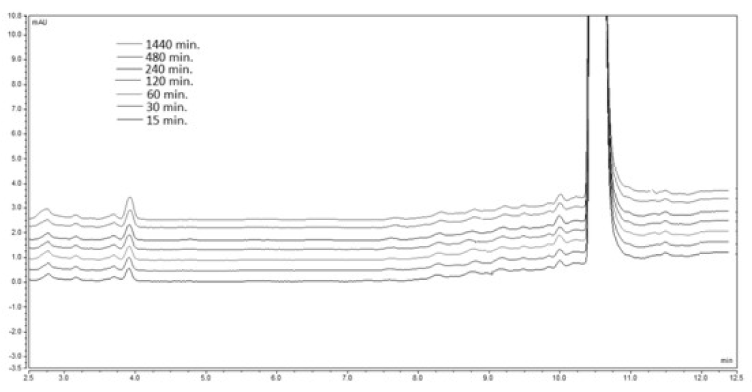
Figure 4. Chromatogram of the analyzed product 1А at рН 1.2 and temperature of 37 ºС.
An appearance of a small peak at the retention time corresponding to 2A (4.0 min.) is observed. In addition a kinetic study of the established degradation was also performed.
Kinetic evaluation of degradation of compound 1А at рН 1.2 (stomach) and temperature of 37 ºС
The corresponding time dependence curve for degree of degradation of compound 1A at pH=1.2 and temperature of 37oC was drawn and presented on Figure 5.
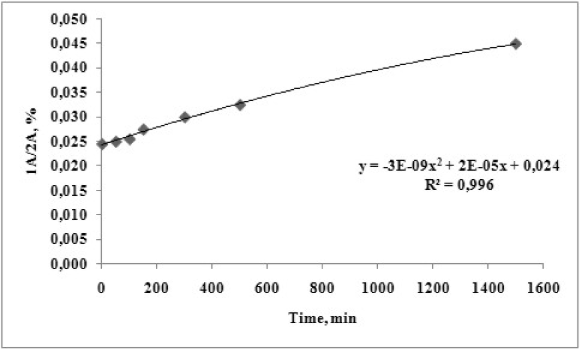
Figure 5. Time dependent degradation of 1А at рН 1.2 and temperature of 37 ºС.
The obtained results reveal about two times accumulation of the hydrolytic product 2A for a 24 h period. A second degree polynomial dependency is observed with R2 of 0.995.
Stability determination of compound 1А at рН 4.5 (urine) and temperature of 37 ºС
Preparation of buffer pH 4.5: A 2 mol/l CH3COOH solution (solution A) is prepared. 0.299 g of CH3COONa and 1.4 ml solution A are mixed and diluted to 100 ml with distilled water.
A 20 mg sample of the model compound was weighed and dissolved in the corresponding mixture of acetonitrile: buffer pH 4.5. The obtained solutions were stirred in a micro reactor at 37oC for a total time of 1440 min. An aliquot 20 μl samples were drawn at the defined above time intervals and injected into the UHPLC system. The obtained chromatogram is presented on Figure 6.
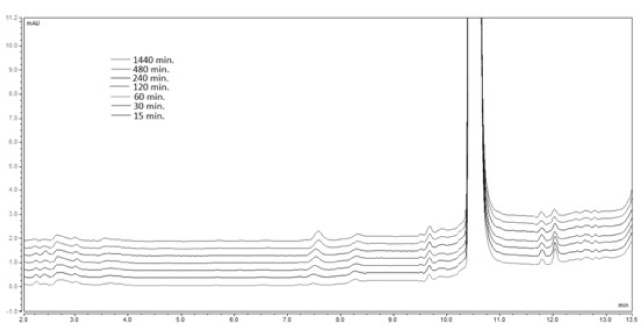
Figure 6. Chromatogram of the analyzed sample 1А at рН 4.5and temperature of 37 ºС.
No new peak, corresponding to the retention time of the degradation product 2A, is formed. The performed kinetic evaluation presented on Figure 7 demonstrates that for the tested period of 1440 min. (24 h) no chemical transformation of the analysed pyrrole ester 1A is observed under the evaluated conditions.
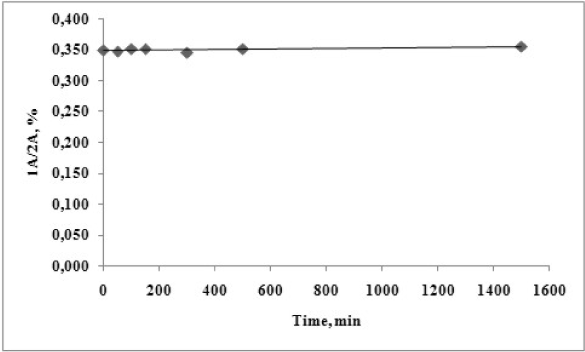
Figure 7. Time dependent degradation of 1А at рН 4.5 and temperature of 37 ºС.
Stability determination of compound 1А at рН 6.8 (blood plasma) and temperature of 37ºС
Preparation of buffer pH 6.8: A mixture of 0.2 mol/l KH2PO4 (solution A) and 0.2 mol/l NaOH (solution B) is prepared, where 25 ml of solution A are mixed with 11.2 ml of solution B. The obtained mixture is diluted to 100 ml with distilled water.
A 20 mg sample of the 2021 Copyright OAT. All rights reservved in the corresponding mixture of acetonitrile: buffer pH 6.8. The obtained solutions were stirred in a micro reactor at 37oC for a total time of 1440 min. An aliquot 20 μl samples were drawn at the defined above time intervals and injected into the UHPLC system. The obtained chromatogram is presented on Figure 8.
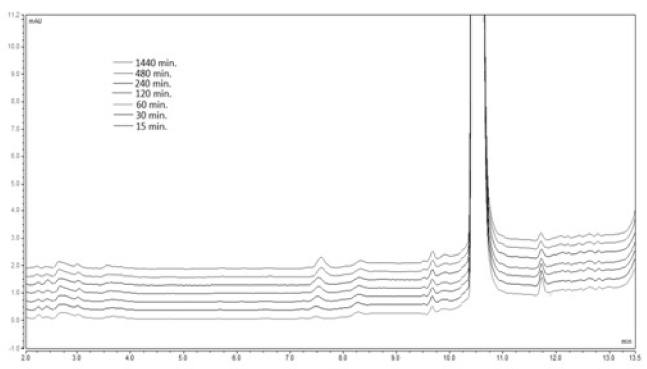
Figure 8. Chromatogram of the analyzed sample 1А at рН 6.8 and temperature of 37 ºС
As seen from the presented chromatogram no new peak, corresponding to the retention time of the degradation product 2A, is formed.
The performed kinetic evaluation presented on figure 9 demonstrates that for the tested period of 1440 min. (24 h) no chemical transformation of the analysed pyrrole ester 1A is observed under the evaluated conditions.
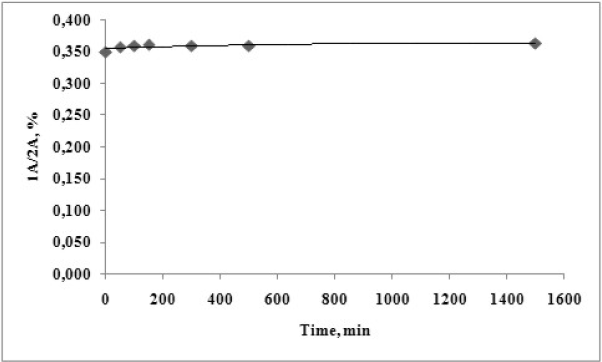
Figure 9. Time dependent degradation of 1А at рН 6.8 and temperature of 37 ºС.
Stability determination of compound 1А at рН 13 and temperature of 37ºС
Preparation of buffer pH 13: A standard buffer with pH 13 (КCl/NaOH) was used for the current study.
A 20 mg sample of the model compound was weighed and dissolved in the corresponding mixture of acetonitrile: buffer pH 13. The obtained solutions were stirred in a micro reactor at 37oC for a total time of 1440 min. An aliquot 20 μl samples were drawn at the defined above time intervals and injected into the UHPLC system. The obtained chromatogram is presented on Figure 10.
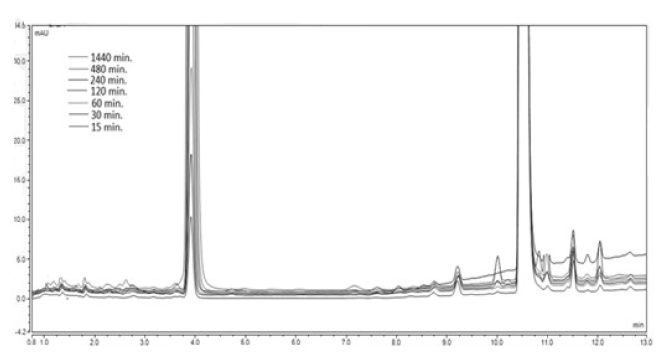
Figure 10. Chromatogram of the analyzed sample 1А at рН 13 and temperature of 37 ºС.
As seen from the presented chromatogram well defined new peaks, appearing at the retention time of the degradation product 2A, are formed, starting from the 15th min of incubation.
Kinetic evaluation of degradation of compound 1А at рН 13 and temperature of 37ºС
The corresponding time dependence curve for degree of degradation of compound 1A at pH = 13 and temperature of 37oC was drawn and presented on Figure 11.
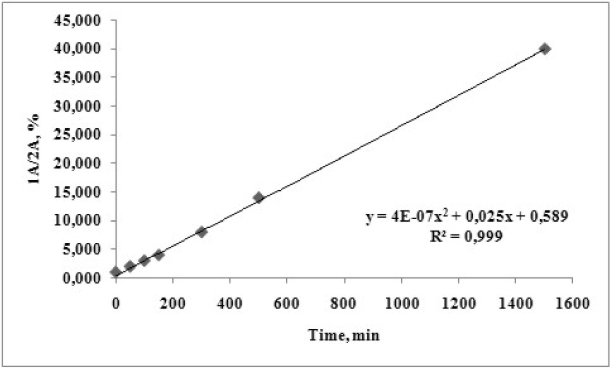
Figure 11. Time dependent degradation of 1А at рН 13 and temperature of 37 ºС.
From the obtained graphical dependency a fast hydrolysis is revealed. A second degree polynomial dependency is observed with R2 of 0.9996. Almost 80-fold increase in the amount of 2A is registered for a 24 h period.
A gradient UHPLC method was used and validated for determination of chemical stability and stability under close to physiological conditions of a pyrrole-containing ester derivative. The proposed method was found to be accurate, precise, reproducible and specific. The results indicate that the tested compound is stable at moderate and low alkali pH and temperatures, but susceptible to hydrolysis in strong acidic (pH 1.2) and strong alkali (pH 13) media. As degradation product the corresponding di-carboxylic acid is obtained.
- Jefford M, Moore R (2008) Improvement of informed consent and the quality of consent documents. Lancet Oncol 9: 485-493. [Crossref]
- Seeking patients' consent: The ethical considerations. London: General Medical Council; 1998.
- Butow PN, Brown RF, Tattersall MH (2000) Ethics of clinical trials. N Engl J Med 342: 978. [Crossref]
- Chandrasekharan DP, Taggart DP (2011) Informed consent for interventions in stable coronary artery disease: problems, etiologies, and solutions. Eur J Cardiothorac Surg 39: 912-917. [Crossref]
- Larobina ME, Merry CJ, Negri JC, Pick AW (2007) Is informed consent in cardiac surgery and percutaneous coronary intervention achievable? ANZ J Surg 77: 530-534. [Crossref]
- Price DD, McGrath PA, Rafii A, Buckingham B (1983) The validation of visual analogue scales as ratio scale measures for chronic and experimental pain. Pain 17: 45-56. [Crossref]
- Childers R, Lipsett PA, Pawlik TM (2009) Informed consent and the surgeon. J Am Coll Surg 208: 627-634. [Crossref]
- Carle C, Ashworth A, Roscoe A (2009) A survey of post-sternotomy chronic pain following cardiac surgery. Anaesthesia 64: 1387. [Crossref]
- Uzzaman MM, Tayeh S, Sinha S, Ratnasingham K, Stoker DL (2011) Consenting practice for laparoscopic cholecystectomy - are we doing enough to warn patients about their operation? Int J Surg 9: 643-647. [Crossref]
- Hoosein MM, Towse H, Conn G, Stoker DL (2008) Consenting practice for open inguinal hernia repairs - are we failing to warn patients of serious complications? Ann R Coll Surg Engl 90: 643-646. [Crossref]