Cancer-specific isoforms (CSIs) have been implicated in cancers. The Cancer Genome Atlas (TCGA) is an interface that helps to systematically investigate disease linkages of CSIs across multiple tumor types of large sample sizes. We analyzed the RNA-Seq datasets of 12 tumor types across different tissues from TCGA. CSIs were identified based on the expression status of a spliced isoform. A total of 693 CSI × tumor-type pairs involving 288 distinct isoforms and 263 distinct genes across 3935 samples of 12 tumor types were analyzed. Out of the 52 known cancer biomarker CSI genes, 48 are being analyzed in clinical trials. Out of 31 CSIs significantly correlated with patient survival time, 18 were from kidney cancer samples. Possible disease linkages in terms of overexpression/amplification ratios of CSI genes were observed in 59% of the CSI × tumor-type pairs. CSIs may therefore be more important than expected in the discovery of cancer drug targets. Few exclusively expressed CSIs could be targeted with high selectivity and safety such as Survivin-2B in lung or kidney cancers.
cancer-specific isoforms, survival time, spliced isoform, cancer genome atlas
Alternative splicing is a regulated process during gene expression that increases the diversity of transcriptome and proteome complexity. It is regulated by a combination of genetic and epigenetic factors such as DNA and histone modifications and mutations within transcriptional factor binding site [1]. Changes in alternative splicing have been implicated in cancers [1]. Many signaling pathways are altered in cancers because of changes in the expression of pathway critical gene isoforms. Different isoforms of a gene may demonstrate opposing biological activity, such as pro- and anti-apoptotic isoforms of caspase 8 (CASP8), tumor suppressors, or oncogenic isoforms of Myc [1]. Previous studies have explored profiles of specific isoforms of a few cancers from different tissues, including prostate, ovarian, colon, bladder, breast, and liver, by using parallel paired-end RNA sequencing or exon arrays [2-8]. Cancer-specific isoforms (CSIs) may provide a pool of candidates for diagnostic markers and therapeutic targets. For example, anti-cancer drugs targeting chronic myeloid leukemia (CML) cell-specific breakpoint cluster region-Abelson (BCR-ABL) fusion proteins can be specially designed for cancer-specific isoforms to minimize toxicity [1]. Moreover, a study by Llpo et al., reported that an prostate specific antigen (PSA) isoform, α2,3-sialic acid was useful in differentiating aggressive and non-aggressive prostate carcinoma and its future implications in translational application in the clinic [9]. Further, it is reported that spliced isoforms of known genes such as DNAJB6 is Involvement of DNAJB6 is implicated in multiple pathologies such as Huntington's disease, Parkinson's diseases, limb-girdle muscular dystrophy, cardiomyocyte hypertrophy and cancer [10]. Wang et al., also reported the role of abnormally expressed micro RNAs and certain transcription factors in prognosis and pathogenesis of osteosarcoma [11].
The primary aim of this study was to comprehensively explore potential disease linkages of CSIs using a publicly available resource. Several reasons warrant this study. First, small sample size has been a major limitation of previously published CSI studies. Second, biologists exploring drug targets need to be aware of the expression profile of isoforms in cancer tissues and their possible relevance to a given disease or safety concern. To date, there are only a few comprehensive, publicly available databases tailored to provide such information. Third, previous works have revealed possible mechanisms of disease linkages only for a few CSIs. To the best of our knowledge, there is no published research systematically exploring disease linkages of spliced isoforms across dozens of tumor types of large sample sizes. In this study, based on our previous finding [12], we systematically revealed splicing signatures from the RNA-Seq datasets of 12 tumor types from different tissues from The Cancer Genome Atlas (TCGA). In total, 693 CSI × tumor-type pairs were chosen across 12 tumor types from 3935 samples. The potential of disease linkages of CSIs was estimated from three different perspectives: known cancer disease biomarkers (37% pairs), possible overexpression/amplification biomarkers (59%), and possible prognostic biomarkers (4%). Some CSIs could be chosen as drug targets when they are exclusively expressed in tumor samples and are absent from normal samples. Although it is not a new concept that specific spliced isoforms are associated with various cancers and that targeting these alterations may be one of the keys to understanding cancers, the significant contribution of our work is the inclusion of CSI candidates of all genes across samples of many tumor types and identification of CSIs that have great potential for cancers.
Use of publicly available datasets in identifying CSIs
Whole transcriptome sequencing, also known as “RNA-Seq,” captures both coding and non-coding RNA in a biological sample. We collected and analyzed data from 3935 RNA-Seq samples. The samples span 12 tumor types from TCGA, which has provided a comprehensive understanding of cancer genomics from different perspectives. The number of samples per tumor type varied between 34 and 849, and their RNA-Seq data, copy number variation data, and clinical data. The 12 tumor types that were collected from different cancer tissues if their RNASeq data of solid tissue normal samples were available: Breast (breast carcinoma BRCA), Endocrine (thyroid carcinoma THCA), Gastrointestinal (liver hepatocellular carcinoma LIHC), Gynecologic (uterine corpus endometrial carcinoma UCEC), Head and Neck (head and neck squamous cell carcinoma HNSC), Thoracic (lung adenocarcinoma LUAD and lung squamous cell carcinoma LUSC) and Urologic (kidney chromophobe KICH, kidney renal cell carcinoma KIRC, kidney renal papillary cell carcinoma KIRP, prostate adenocarcinoma PRAD, and bladder carcinoma BLCA).
The study protocol was approved by R&D information China, AstraZeneca, and the study was strictly conducted as per STREGA guidelines.
Identifying CSIs from RNA-Seq datasets
The method used to identify CSIs from RNA-Seq datasets is described in Figure 8A and 8B. The expression status of spliced isoforms was determined by the method discussed elsewhere [9]. First, the expression status of all the spliced isoforms of a tumor sample was determined from their expression values of specific exon sets or junction sets. The expression status of all the spliced isoforms of pooled solid tissue normal samples was determined in the same manner from the averaged expression values of specific exons or junctions. Second, CSI candidates of a tumor sample were determined by either of two criteria: 1) if an isoform was found to be expressed in a tumor sample but absent in the solid tissue normal samples and 2) if an isoform was expressed in both a tumor sample and the solid tissue normal samples, only if the fold change of averaged expression values of all specific exons or junctions of the isoform between the tumor sample and the solid tissue samples was greater than five. Third, CSI candidates of tumor types were determined. The final set of CSIs included isoforms whose occurrence ratios in a tumor type were no less than 5%.
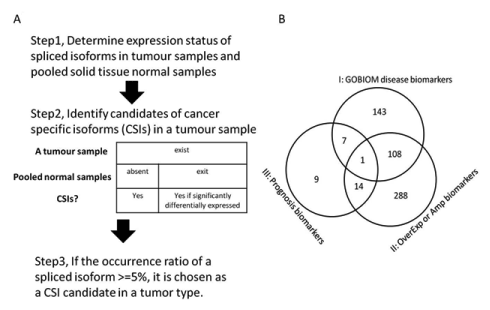
Figure 8.Pipeline for finding cancer-specific isoforms from RNA-Seq datasets and the CSI functional annotation
(A) Description of the method for finding cancer-specific isoforms from RNA-Seq datasets; (B) The overlapping profile of three classes of CSI × tumor-type pairs. Class I are made up of CSI × tumor-type pairs in which CSI genes are GOBIOM disease biomarkers. Class II are made up of CSI × tumor-type pairs in which CSI genes are significantly overexpressed or amplified in matched tumor types. Class III is CSI × tumor type pairs in which CSIs may be prognostic biomarkers for the matched tumor types.
Statistical methods
CSIs that significantly correlated with patient survival time were identified by two steps. First, Cox proportional hazards model was used, in which each CSI was respectively included and adjusted by other clinical parameters, including clinical stage, age and sex, and P<0.05 was assumed to be significant. This procedure was performed by using the survival package in R (version 3.1) project. Second, the following empirical criteria were used to filter data: 1) overall survival ratios of the two groups must be no greater than 3% and 2) the sample sizes of the two patient groups with or without CSI must be extremely unbalanced, and the chosen candidates were further analyzed by Kaplan-Meier method.
Summary of CSIs across TCGA tumor types
A total of 693 CSI × tumor-type pairs across 12 tumor types were identified in the present analysis. These 693 pairs involved 288 distinct isoforms and 263 distinct genes. The number of isoforms and their occurrence ratios per tumor type varied considerably. In terms of CSI count per tumor type, some tumor types had about a hundred CSIs such as KIRP, LUCS/LUAD, and LIHC. Some tumor types had a sizable number of CSIs such as THCA and PRAD. In terms of the occurrence ratio in a tumor type, most pairs involved 5%-10% patients; however, some tumor types were enriched with CSIs of high occurrence ratio (≥ 20%) such as LUCS, KIRP, and LUAD (Figure 1). Moreover, 124 CSIs were unique to a specific tumor type and 164 CSIs were shared by two or more tumor types. CSIs of greater tumor type counts were more likely to have high occurrence ratios (Figure 2).
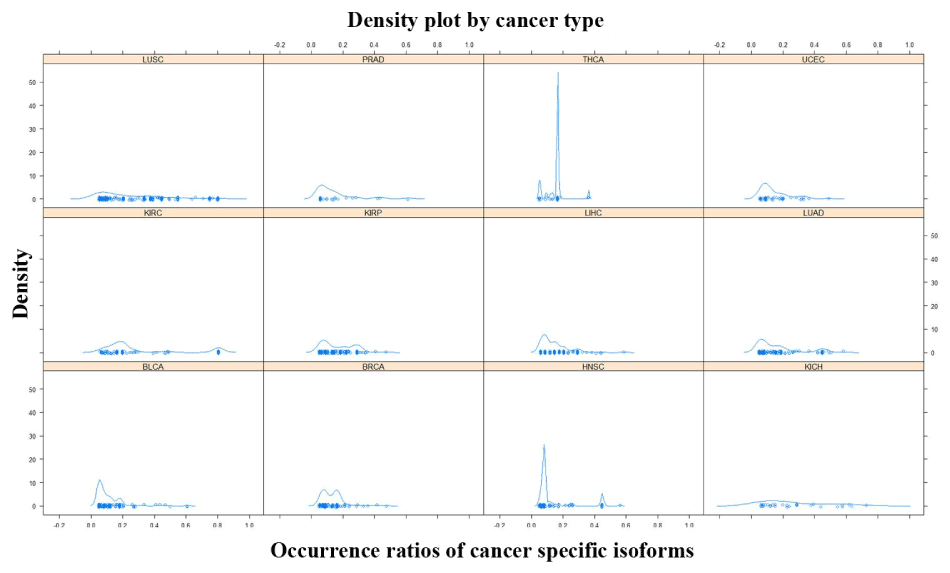
Figure 1.The density plot of occurrence ratios of cancer specific isoforms across tumor types
The X-axis represents the occurrence ratios of cancer specific isoforms in a tumor type. The Y-axis represents the density of the corresponding occurrence ratios.
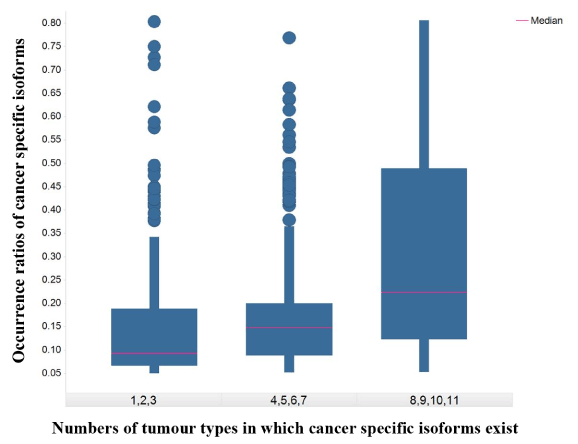
Figure 2.The box plot of occurrence ratios of cancer specific isoforms against numbers of tumor types in which cancer specific isoforms exist
The X-axis represents numbers of tumor types in which cancer specific isoforms exist. The Y-axis represents the occurrence ratios of cancer specific isoforms in a tumor type.
Biological functions of CSIs are explored in terms of the biological processes they involve in. As shown Figure 3, CSI genes are mapped to 63 Gene Ontology (GO) terms, which mainly belong to “metabolic process,” “response to stimulus,” “cell communication,” “cell growth & death,” “biosynthetic process,” “transcription,” “transport,” and “protein modification [13]. A GO term is scored by the average of occurrence ratios of genes mapped to it. Although almost all GO terms exist in all the tumor types, scores of GO terms are different across tumor types (Figure 3). For example, the scores of GO terms in LUSC and KICH or KIRC are always greater than LUAD and KIRP, respectively. It means the distribution of occurrence ratios of CSIs across tumor types is still uneven even when they are grouped by GO terms. The enrichment and clustering analysis of CSI genes by using the online tool Database for Annotation, Visualization and Integrated Discovery (DAVID) is shown in Supplementary Table file [14]. The top clusters with high enrichment scores contain biological processes such as cell death and cell cycle.
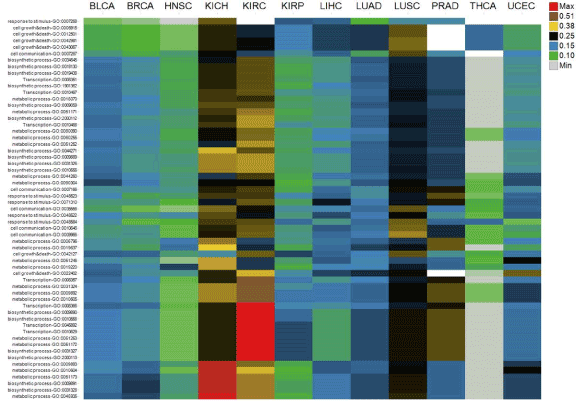
Figure 3.Biological functions of cancer specific isoforms across tumor types. The columns are tumor types
The rows are Gene Ontology terms of genes of cancer specific isoforms. Cell values are the average of occurrence ratios of all the cancer specific isoforms whose corresponding genes are mapped to Gene Ontology terms. Different cell value ranges are indicated by different colors.
Known disease biomarkers of CSI genes
We collected all genes categorized by disease biomarkers for different tumor types from GVK BIO Online Biomarker Database (GOBIOM), a comprehensive biomarker database that gathers information from clinical trials, scientific conferences, regulatory-approved documents, literature databases, and so on.
Of the 263 CSI genes, 52 were found in GOBIOM cancer disease biomarkers. In terms of pharmaceutical R&D pipeline phases of disease biomarkers, 40 CSI genes are in clinical trials (2 in phase IV, 8 in phase III, 15 in phase II, and 15 in phase I) and other CSI genes are in the preclinical or exploratory phase. A CSI gene involved in different GOBIOM diseases may be in different pipeline phases. The percentage of CSI genes annotated as cancer disease biomarkers per tumor type and functional category varies between 7%-39% and 29%-50%, respectively.
A gene can be annotated as a disease biomarker for multiple tumor types in GOBIOM. The more the GOBIOM tumor types, the more is the possibility of CSI genes having strong disease linkages with cancers. As shown in Figure 4, there were 31 CSI genes whose numbers of GOBIOM tumor types were greater than 1. A CSI gene is annotated differently between GOBIOM and TCGA tumor types, and if one of the TCGA tumor types of the CSI gene is also found in the GOBIOM tumor types, the disease linkage of that CSI gene and the TCGA tumor type is classified as “validated.” Of the 52 CSI genes, 22 had “validated” disease linkages with some cancers (Figure 4).
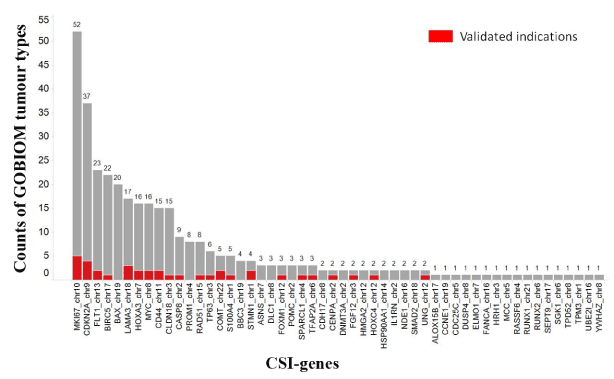
Figure 4.The existence of CSI genes across GOBIOM tumor types
The color Red indicates the overlap of GOBIOM and TCGA tumor types. The X-axis represents the genes of cancer specific isoforms. The Y-axis represents the number of GOBIOM tumor types in which a CSI gene exists.
The occurrence ratio of CSIs per tumor type and the TCGA tumor type count of CSIs are two simple and direct measurements for disease linkages of CSIs. With an increase in the occurrence ratio of CSIs, the proportion of CSI genes that was annotated as disease biomarkers increased. For example, more than half of the CSI genes whose occurrence ratios were greater than 50% had evidence of disease linkages with some tumor types. The same trend was observed for the TCGA tumor type count of CSIs. With an increase in the tumor type count, the proportion of CSI genes that was annotated as disease biomarkers increased.
Correlations of CSIs with clinical phenotypes
A common strategy for identifying cancer drivers is to associate genomic alterations with clinical phenotypes. Some CSIs were observed to be significantly correlated with patient survival times. They were considered as candidates of prognosis biomarkers and termed prognosis CSIs. In total, 31 prognosis CSIs (Figure 5; Supplementary Table 2) were involved in different tumor types and biological functional categories. Of the 32 prognosis CSIs, 8 were annotated as GOBIOM disease biomarkers. According to the hazard ratio calculations taken from two groups of patients – those with or without a CSI – prognosis CSIs were classified into two groups: poor prognosis biomarkers and good prognosis biomarkers.
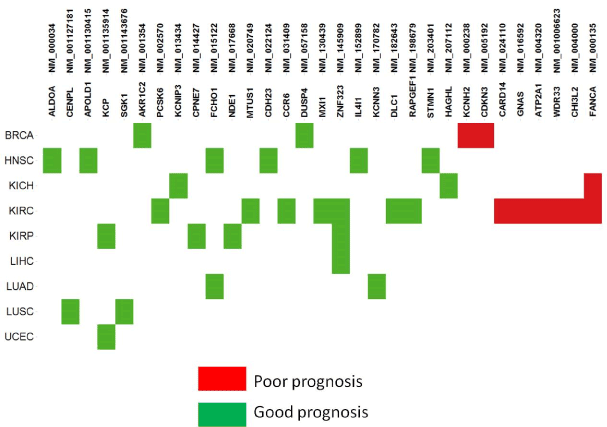
Figure 5.The prognostic biomarkers of cancer specific isoforms across tumor types
Most of the poor prognosis CSIs were found to be enriched with tumor type KIRC. NM_000238 is the longest isoform of KCNH2 (Potassium Channel, Voltage Gated Eag Related Subfamily H, Member 2) that encodes a protein known as Kv11.1 (ether-a-go-go related gene/ Herg), the alpha subunit of voltage-gated potassium channels. Overexpression of Kv11.1 resulted in poor prognoses of ovarian and stomach cancer [15,16]. NM_016592 encodes neuroendocrine secretory protein 55 (NESP55), which is expressed in neuroendocrine tumors of the pancreas and adrenal glands [17]. It is an isoform of GNAS encoding the guanine nucleotide-binding protein (G-protein) alpha subunit. GNAS mutations are specific to intraductal papillary mucinous neoplasms with a poor prognosis, a subtype of pancreatic cystic neoplasm. Another GNAS splice variant may contribute to the pathogenesis of estrogen receptor-positive breast cancer, which is linked to poor prognosis [18]. NM_004000 encodes the longest isoform of chitinase 3-like 2 (CHI3L2). The CHI3L1 and CHI3L2 genes of chitinase-like cartilage protein have a high homology of nucleotide and amino acid sequences. Either CHI3L1 or CHI3L2 activates the PI3K/AKT pathway and a MAP kinase signaling cascade [19-21]. Their overexpression was significantly correlated with poor survival in glioblastoma [22].
Good prognosis CSIs significantly associated with prolonged survival times were found to be distributed across multiple tumor types. Although they could probably be true prognosis biomarkers for some tumor types, it is difficult to determine if they are also cancer drivers. With limited scope of our knowledge, we cite one example to show that the class of prognosis CSIs may be of importance in cancer research. NM_130439 (MXI1-SRalpha) encodes a splicing isoform of gene MXI1 (MAX-interacting protein 1), which is a member of the Myc family of transcriptional regulators. The overexpression of MXI1 was significantly correlated with a better disease-specific survival in women with breast cancer in China [23]. MXI1 methylation is one of the high-risk biomarkers of medulloblastoma [24]. In addition, MXI1 overexpression in primary human clear cell kidney cancers reprograms cellular energy metabolism and protects cells from c-Myc–dependent apoptosis [25,26].
Overexpression and amplification profiles of CSI genes
Because genes necessary for tumorigenesis, proliferation, and metastasis are always amplified or overexpressed, DNA amplification or gene overexpression can be chosen as a biomarker to estimate the linkage potential of the gene to the respective cancer. Of note, genes that are both overexpressed and amplified to a high ratio in a tumor type usually act as drug targets. We assume that if a gene has a linkage with a tumor type, its CSIs may also have the same disease linkages.
The degree of overexpression or amplification ratios of CSI genes in a tumor type allows for estimation of the likelihood of CSIs having disease linkages with the same tumor types. The overexpression ratios of 194 CSI gene × tumor type were greater than 10% (Figure 6). The density plots of the overexpression ratios of the three classes of CSI genes classified by pharmaceutical R&D pipeline phases were similar. Moreover, there was no linear relationship between CSI-gene overexpression ratios and CSI ratios; that is, genes with small gene-overexpression ratios could have large CSI sample ratios and vice versa. Therefore, the expression of CSIs could sometimes be better biomarkers than the expression of its genes. For example, NM_000610 (CD44v6) encoding the longest transcript of CD44 existed in 38% of the LUSC samples, but the overexpression ratio of CD44 in LUSC was zero. CD44v6 was expressed in lung squamous cell carcinomas and significantly correlated with lymph node metastases and tumor size [27].
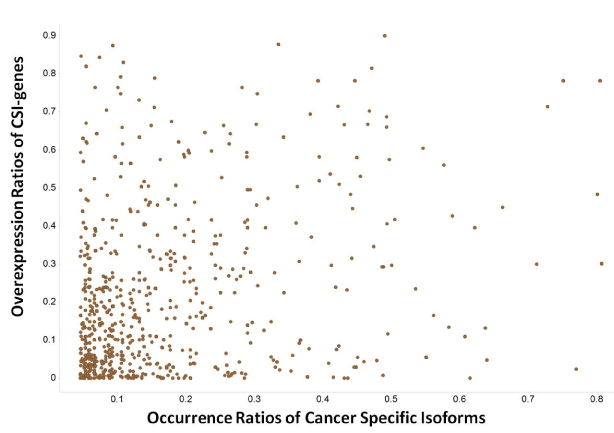
Figure 6.The profile of overexpression ratios of genes of cancer specific isoforms (CSI-genes)
The X-axis represents the occurrence ratios of cancer specific isoforms. The Y-axis represents the overexpression ratios of genes of cancer specific isoforms.
The amplification ratios of 27 CSI genes × tumor types were greater than 5%, signifying that the genes of CSIs are less likely to be amplified. CSI genes that were both overexpressed and amplified are shown in Figure 7. Although only some CSI genes are annotated as GOBIOM disease biomarkers, others may also have potential for disease linkages. For example, RAB3IP, the human homolog of an interactor of the Ras-like GTPase Rab3A, was overexpressed and amplified in three cancer types: BLCA), LUAD, and head and neck squamous cell carcinoma HNSC. In the three cancer types, sample ratios of a splice variant of RAB31P, NM_00102467, were 28%, 26%, and 17%, respectively. To the best of our knowledge, there has been no link established between RAB31P and cancers; however, its interaction protein Rab3A was shown to be involved in tumorigenesis [28,29].
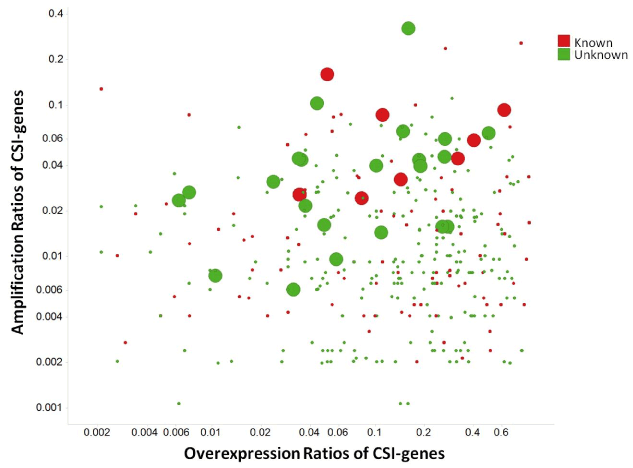
Figure 7.The scatter plot of amplification ratios and overexpression ratios of cancer specific isoform genes (CSI-genes)
Large dots are CSI genes which have significant amplification and overexpression correlations. 'Known' stands for CSI genes annotated as GOBIOM cancer disease biomarkers.
Query interface
An interface is offered for querying and downloading all the detailed data in this study. It can be used to explore either patient-level data (specific exon/junctions of genes and their expression values, the existence status of isoforms, and so on) or disease-level data (the occurrence ratios of CSIs and corresponding gene overexpression/amplification ratios). The interface is available online at http://dev-rwe.rwebox.com/TCGA/ (Accessed on June 20, 2016) [30].
We analyzed 693 CSI × tumor-type pairs identified from the RNA-Seq datasets of 12 TCGA tumor types. These pairs were classified into three classes. Class I had 255 pairs (37%) whose CSI genes were GOBIOM cancer disease biomarkers. Class II had 411 (59%) pairs whose CSI genes were either overexpressed or amplified in matched tumor types. Class III had 31 pairs (4%) whose CSIs were possible prognostic biomarkers. Class IV had 188 pairs (27%) whose disease linkages are to be further explored. Of the Class II pairs, 27% were also in Class I, signifying the presence of alternative spliced isoforms apart from overexpression or amplification, which may be a new mechanism by which those genes contribute to cancers. Of the Class III pairs, 58% were not found in Class I but were found in Class II, signifying that these CSI genes may be novel cancer genes, although this requires further confirmation. The only CSI shared by Classes I-III was NM_000135 of KIRC, which encodes the longest isoform of FANCA (Fanconi anaemia, complementation group A) gene. To the best of our knowledge, there is no published research on the linkage of this gene with kidney renal cell cancer. Out of the 52 known cancer biomarker CSI genes, 48 have already been used in clinical trials; disease linkages of 22 CSI genes with matched tumor types were discovered in previous studies. Out of 31 CSIs significantly correlated with patient survival time, 18 were found in kidney cancers. This signifies that few alternative spliced isoforms may be key drivers of kidney cancers, especially kidney renal cell carcinoma.
Some isoforms are not included in this work for two reasons. First, this work was based on known genomic annotation structures and therefore few novel isoforms were missed out. Second, we mined CSI candidates in terms of a single isoform level. As a result, some well-known isoform switching events may have been missed because they were involved in multiple isoforms. For example, reported tumor-specific isoforms switch between FGFR2-IIIb (NM_001144919, NM_022970) and FGFR2-IIIc (NM_001144915, NM_001144916, NM_001144918, NM_000141) of the fibroblast growth factor receptor 2 (FGFR2) in kidney cancers were not included in this study [31].
Some CSIs may be specifically targeted by compounds, antibodies, or siRNA with high selectivity when they meet one or more of the following criteria: 1) they are druggable; 2) they are high-expressed membrane proteins for antibodies; 3) they are enzymes or kinases for compounds; or 4) they are exclusively expressed in tumor samples for high efficacy and safety. For example, in our study, NM_001012271 (Survivin-2B), a splice variant of BIRC5 (Baculoviral IAP Repeat Containing 5), was found to be exclusively expressed in tumor samples of KIRP, LUAD, and LUSC, with high occurrence ratios of 29%, 19%, and 34%, respectively, and not expressed in adjacent solid tissue normal samples. A study by Zhang et al. [32], reported that multiple genes expression was associated with adverse overall survival and relapse-free survival (RFS) in non-small cell and small cell lung carcinoma. The anti-apoptotic role of and the high expression of Survivin-2B were correlated with a lower survival ratio of patients with acute myeloid leukemia (AML) in vitro [33,34]. Targeting Survivin-2B could be an effective means for patients with KIRP, LUAD, or LUSC to overcome their resistance to standard treatment regimens such as chemotherapy. The drug potential and possible disease linkages of other CSI candidates warrant investigation and validation by oncology researchers.
The authors declare that they have no competing interests.
Jia Wei conceived this study; Yan Ji performed analysis; Ming Chen developed the web application; Yan Ji, Yunqin Chen and Jia Wei participated in writing the manuscript. All authors have read and approved the final version of the manuscript.
We appreciate Ziliang Qian and Dong Hua who gave us some useful suggestions for this work.
- Pal S, Gupta R, Davuluri RV (2012) Alternative transcription and alternative splicing in cancer. Pharmacol Ther 136: 283-294. [Crossref]
- Zhang C, Li HR, Fan JB, Wang-Rodriguez J, Downs T, et al. (2006) Profiling alternatively spliced mRNA isoforms for prostate cancer classification. BMC Bioinformatics 7: 202. [Crossref]
- Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Gervais-Bird J, et al. (2008) Multiple alternative splicing markers for ovarian cancer. Cancer Res 68: 657-663. [Crossref]
- Thorsen K, Sorensen KD, Brems-Eskildsen AS, Modin C, Gaustadnes M, et al. (2008) Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol Cell Proteomics 7: 1214-1224. [Crossref]
- Venables JP, Klinck2021 Copyright OAT. All rights reservin G, et al. (2008) Identification of alternative splicing markers for breast cancer. Cancer Res 68: 9525-9531. [Crossref]
- Xi L, Feber A, Gupta V, Wu M, Bergemann AD, et al. (2008) Whole genome exon arrays identify differential expression of alternatively spliced, cancer-related genes in lung cancer. Nucleic Acids Res 36: 6535-6547. [Crossref]
- Huang Q, Lin B, Liu H, Ma X, Mo F, et al. (2011) RNA-Seq analyses generate comprehensive transcriptomic landscape and reveal complex transcript patterns in hepatocellular carcinoma. PloS one 6: e26168. [Crossref]
- Misquitta-Ali CM, Cheng E, O'Hanlon D, Liu N, McGlade CJ, et al. (2011) Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol 31: 138-150. [Crossref]
- Llop E, Ferrer-Batallé M, Barrabés S, Guerrero PE, Ramírez M, et al. (2016) Improvement of Prostate Cancer Diagnosis by Detecting PSA Glycosylation-Specific Changes. Theranostics 6: 1190-1204. [Crossref]
- Meng E, Shevde LA, Samant RS, et al. (2016) Emerging roles and underlying molecular mechanisms of DNAJB6 in cancer. Oncotarget. [Crossref]
- Wang T, Xu Z, Wang K, Wang N (2015) Network analysis of microRNAs and genes in human osteosarcoma. Exp Ther Med 10: 1507-1514. [Crossref]
- Yan Ji JW (2013) Isoform Inference From RNA-Seq Samples Based on Gene Structures on Chromosomes. Journal of Biosciences and Medicines 1: 1-5.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25-29. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57. [Crossref]
- Cicek MS, Koestler DC, Fridley BL, Kalli KR, Armasu SM, et al. (2013) Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum Mol Genet 22: 3038-3047. [Crossref]
- Ding XW, Yang WB, Gao S, Wang W, Li Z, et al. (2010) Prognostic significance of hERG1 expression in gastric cancer. Dig Dis Sci 55: 1004-1010. [Crossref]
- Jakobsen AM, Ahlman H, Keby L, Abrahamsson J, Fischer-Colbrie R, et al. (2003) NESP55, a novel chromogranin-like peptide, is expressed in endocrine tumours of the pancreas and adrenal medulla but not in ileal carcinoids. Br J Cancer 88: 1746-1754. [Crossref]
- Garcia-Murillas I, Sharpe R, Pearson A, Campbell J, Natrajan R, et al. (2014) An siRNA screen identifies the GNAS locus as a driver in 20q amplified breast cancer. Oncogene 33: 2478-2486. [Crossref]
- Areshkov PO, Avdieiev SS, Iershov AV, Kavsan VM (2011) Stimulation of transient versus sustained ERK1/2 phosphorylation by relative chitinase-like proteins CHI3L1 and CHI3L2 correlates with different kinase localization and biological outcome. Biopolymers and Cell 27: 343-346.
- Areshkov PA, Kavsan VM (2010) Chitinase 3-like protein 2 (CHI3L, YKL-39) activates phosphorylation of extracellular signal-regulated kinases ERK1/ERK2 in human embryonic kidney (HEK293) and human glioblastoma (U87 MG) cells. Tsitol Genet 44: 3-9. [Crossref]
- Dmitrenko VV, Avdieiev SS, Areshkov PO, Balynska OV, Bukreieva TV, et al. (2013) From reverse transcription to human brain tumors. Biopolymers and Cell 29: 221-233.
- Saidi A, Javerzat S, Bellahcee, De Vos J, Bello L, et al. (2008) Experimental anti-angiogenesis causes upregulation of genes associated with poor survival in glioblastoma. Int J Cancer 122: 2187-2198. [Crossref]
- Xu LP, Sun Y, Li W, Mai L, Guo YJ, et al. (2014) MYC and MXI1 protein expression: potential prognostic significance in women with breast cancer in China. Oncol Res Treat 37: 118-123. [Crossref]
- Schwalbe EC, Williamson D, Lindsey JC, Hamilton D, Ryan SL, et al. (2013) DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125: 359-371. [Crossref]
- Tsao CC, Teh BT, Jonasch E, Shreiber-Agus N, Efstathiou E, et al. (2008) Inhibition of Mxi1 suppresses HIF-2a-dependent renal cancer tumorigenesis. Cancer Biol Ther 7: 1619-1627. [Crossref]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, et al. (2007) HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11: 407-420. [Crossref]
- Afify AM, Tate S, Durbin-Johnson B, Rocke DM, Konia T (2011) Expression of CD44s and CD44v6 in lung cancer and their correlation with prognostic factors. Int J Biol Markers 26: 50-57. [Crossref]
- Lodhi SS, Farmer R, Singh AK, Jaiswal YK, Wadhwa G (2014) 3D structure generation, virtual screening and docking of human Ras-associated binding (Rab3A) protein involved in tumourigenesis. Mol Biol Rep 41: 3951-3959. [Crossref]
- Kim JK, Lee SY, Park CW, Park SH, Yin J, et al. (2014) Rab3a promotes brain tumor initiation and progression. Mol Biol Rep 41: 5903-5911. [Crossref]
- https://dev-rwe.rwebox.com/TCGA/ (Accessed on June , 2016)
- Zhao Q, Caballero OL, Davis ID, Jonasch E, Tamboli P, et al. (2013) Tumor-specific isoform switch of the fibroblast growth factor receptor 2 underlies the mesenchymal and malignant phenotypes of clear cell renal cell carcinomas. Clin Cancer Res 19: 2460-2472. [Crossref]
- Zhang C, Min L, Zhang L, Ma Y, Yang Y, et al. (2016) Combined analysis identifies six genes correlated with augmented malignancy from non-small cell to small cell lung cancer. Tumour Biol 37: 2193-2207. [Crossref]
- Shi K, An J, Shan L, Jiang Q, Li F, et al. (2014) Survivin-2B promotes autophagy by accumulating IKK alpha in the nucleus of selenite-treated NB4 cells. Cell Death Dis 5: e1071. [Crossref]
- Wagner M, Schmelz K, Wuchter C, Ludwig WD, Dorken B, et al. (2006) In vivo expression of survivin and its splice variant survivin-2B: impact on clinical outcome in acute myeloid leukemia. Int J Cancer 119: 1291-1297. [Crossref]