Abstract
((E)-2-(2-hydroxybenzylideneamino)phenolato-2,2-diphenyl-6-aza-1,3-dioxa-2-stanna-[d,h]dibenzocyclononene, [Sn(Ph2SB)] (compound 1, where Ph2SB=(E)-2-(2-hydroxybenzylideneamino)phenolato Schiff base) and two novel compounds, [[SnPh2(F-azoSB)] (compound 2, where F-azoSB=4-((E)-(4-fluorophenyl)diazenyl)-2-((E)-(2-hydroxyphenylimino)methyl)phenolato Schiff base), [[SnPh2(sulf-azoSB)]0.125CHCl3 (compound 3, where sulfamerazineazosalSB=4-((E)-(4-hydroxy-3-((E)-(2-hydroxyphenylimino)methyl)phenyl)diazenyl)-N-(4-methylpyrimidin-2-yl) benzenesulfonamide Schiff base), and the control compound, cisplatin (compound 4) were analysed to comparatively determine their effect on cancer cell growth. Anti-cancer properties of compounds 1-4 were examined using glioblastoma (U-1242 MG), colorectal (HT-29 and HCT-116), and skin (A431) human cancer cell lines. With regards to human glioblastoma cells, compounds 1 and 3 demonstrated anti-proliferative capacity in the cell line tested. Specifically, compounds 1 and 3 inhibited cell proliferation by 50% at concentrations between 10 and 50 µM. With respect to colon cancer cell lines, the IC50 values for compounds 1-3 ranged from 3.04 ± 0.98 to 104.51 ± 13.87 mM. In the case of HCT116, this translates to a 3- to 73-fold inhibitory effect of compounds 1-3 over cisplatin. In all cell lines tested, the chemo-effect was more pronounced with compounds 1-3 than with the control (compound 4); demonstrating that these azo-containing Sn(IV) complexes were more potent than compound 4. The overall effect of compounds 1-3 in the induction of appotosis and the inhibition of proliferation have defined an essential role for these compounds in chemotherapy.
Compound 1
Key words
Sn(IV) complexes, 119Sn NMR spectroscopy, cancer, X-ray crystallography, chemotherapy
Introduction
From the late 1960s until the present, there have been significant advances in both the early detection and the treatments of cancer. Despite these advances overall incidences of cancer and deaths from cancer have increased during the same period [1]. These counterintuitive increases have been the impetus for development of new drugs for cancer treatment. In this context the organometallic compounds have been widely investigated as potential anti-tumour agents. Metallocene dihalide complexes Cp2MX2 (where M=titanium, vanadium, niobium, or molybdenum) were the first early transition metal complexes that were shown to have anti-tumour activity. In addition, the organometallic-DNA and organometallic-nucleic acid interactions of these compounds have also been investigated [2-4]. More recently, ferricenium salts, organotin, and bismuth complexes have also emerged as examples of organometallic compounds that have been found to exhibit interesting anti-tumour activity [5].
The biological activity of organotin compounds has become well known due to their practical applications as fungicides, bactericides, biocides, and pesticides [3,6-8]. It is now well established that organotin compounds are very important in cancer chemotherapy [9] and as potential anti-cancer agents [10-13]. This is due in part to their apoptotic inducing character [14-17]. Recent studies [18-20] have shown that low doses of organotins exhibit anti-tumour activity and suggest an action mode via a gene-mediated pathway in the cancer cells, opening a new research sub-area on organotin compounds. Since there are two primary modes of apoptosis (extrinsic and intrinsic), metal-induced apoptosis is thought to be initiated intracellularly, the mitochondria being most pertinent in mediating apoptosis via metal-induced reactive oxygen species [21]. Di-n-butyltin and tri-n-butyltin chloride, with these organotins, are known to induce apoptosis in vitro in rat thymocytes; inhibiting DNA synthesis and increasing RNA synthesis [22]. More recently, Schiff bases and their tin and organotin derivatives [23-28], tested in vitro in anti-tumour assays, showed moderate-good effect. However, di-n-butyl-tin(IV) compounds in concert with ortho-aminophenols demonstrated better anti-tumour activities while wielding less damage in normal cells as compared to cisplatin [29]. It is believed that di-phenyltin(IV) derivatives may demonstrate efficacy with less toxicity than other di-organo-tin(IV) derivatives due to their interaction with the cell membrane [30-33].
Additional biological studies of a series of organotin(IV) carboxylates (i.e., [Bu2SnL2], [Et2SnL2], [Me2SnL2], [Bu3SnL]n, [Me6Sn2L2]n, [Ph3SnL]n, and [Oct2SnL2] (where L=O2CCH2C6H4OCH3-4)) [34], have been reported. Bioassay results have shown these compounds to have good anti-bacterial, anti-fungal, and anti-tumour activity [34]. Of interest to this work, the effect of these compounds against PC-3 prostate cancer cell line decreased in the following order: [Bu2SnL2] > [Me6Sn2L2]n >[Et2SnL2] > [Me2SnL2] > [Oct2SnL2] [34]. There have also been reports of biological studies involving novel neutral and cationic organotin complexes with pyruvic acid thiosemicarbazone, H2pt, [SnPh2(pt)], [SnMe2(Hpt)(H2O)]Cl, and [SnPh2(Hpt)(H2O)]Cl [35]. The in vitro cytotoxic activity with each complex was evaluated against the cells of three human cancer cell lines: MCF-7 (human breast cancer cell line), T-24 (bladder cancer cell line), A-549 (non-small cell lung carcinoma) and a mouse L-929 (a fibroblast-like cell line cloned from strain L) [35]. [SnPh2(pt)] was found to be the most active of the complexes tested. These compounds generated IC50 values in the μM range; equal to or lower than that of cisplatin, our control compound [34]. Thus, the cytotoxic activity shown by these compounds against each cancer cell line suggested that coupling of H2pt with R2Sn(IV) metal centre resulted in complexes with important biological properties and remarkable cytotoxic activity. In accordance, [SnPh2(pt)] was considered an agent with potential anti-tumour activity and was projected to be a candidate for further in vitro and/or in vivo screening [35].
Seven di-phenyltin(IV) compounds [36] were assessed as new anti-cancer agents against six human tumour cell lines U251, PC-3, K-562, HCT-15, MCF-7 and SKLU-1 [36]. The results suggested biological specificity towards U251, MCF-7 and SKLU-1 cancer cells at doses below 2.5 μM; which are lower than the IC50 value of cisplatin. Since the inhibitory concentration values for the series were similar to Ph2SnCl2, it was concluded that only the Ph2Sn moiety was responsible for those activities [36].
In recent years, our laboratory has been interested in the biological studies of azo substituted salicylaldehyde, its derivatives and coordination complexes synthesised from such ligands. In this study, we have synthesised two novel azo-containing pentacoordinated organotin complex derived from 2-aminophenol. In accordance with previous studies, the aim of this study is to evaluate the in vitro effect (cytotoxicity) of these novel compounds against five human tumour cell lines. This is accomplished by comparing the efficacies of these compounds with the known non-azo containing complex 2,2-diphenyl-6-aza-1,3-dioxa-2-stanna-[d,h]dibenzocyclononene (compound 1) as reported by Beltrán and co-workers [36].
Materials and methods
Reagents
Analytical or reagent grade chemicals were used throughout this study. All the chemicals including solvents were obtained from Sigma-Aldrich (St. Louis, MO, USA) or other commercial vendors, and used as received. Microanalyses (C, H, N) were performed by Desert Analytics, Tucson, U.S.A. and Columbia Analytical Services 3860 S. Palo Verde Road Suite 303 Tucson, AZ 85714, U.S.A. 1H NMR spectra were recorded in CDCl3 on a Varian 500 MHz spectrometer operating at room temperature. 19F and 119Sn NMR spectra were acquired on a Varian 500 MHz spectrometer with CDCl3 as solvent, with CF3CO2H and Me4Sn [36] external references, respectively.
FT IR were acquired in the range 4000-400 cm-1 using the ATR accessory (with a diamond crystal) on a Nicolet 6700 FTIR spectrophotometer. Electronic spectra were recorded using quartz cuvettes on a HP8452 diode array spectrophotometer using DMSO as the solvent. Fluorescence spectra were recorded on a Cary Eclipse fluorescence spectrophotometer (Varian Inc.) using a slit width of 10 nm. ESI MS was carried out on an HP Agilent 1956b single-quadrupole mass spectrometer. Samples were dissolved in either pure methanol or an acetic acid/methanol mixture; then the solution was introduced by direct injection using a syringe pump with a flow rate of 100 mL s-1, while sweeping the cone voltage from 0 to 200 V at a rate of 10 V min-1.
100 mM stock solutions of compounds 1-3 and cisplatin were prepared in DMSO; with the final DMSO concentration in all media was adjusted to 1%.
X-ray crystallography
X-ray intensity data were measured at low temperature with graphite-monochromated radiation (l=0.71073 A°) on a Rigaku AFC8S diffractometer equipped with a 1 K Mercury CCD detector. The structure was solved using direct methods and refinement was done using full matrix least-squares techniques (on F2) [37,38]. Data were corrected for absorption, using semi-empirical methods. Structure solution, refinement and the calculation of derived results were performed with the SHELXTL package of computer programs [39]. One of the coordinated phenyl rings (C26-C31) shows significant disorder and was refined as two slightly displaced rings with each having 50% occupancy.
Crystallographic data for compound 2 has been deposited at Cambridge Crystallographic Data Centre as supplementary publication number CCDC No.=1430652. Copies of available material can be obtained on application to CCDC, 12 Union Road, Cambridge CB2 1Ez, UK (fax: 044 1223336 033 or e-mail: deposit@ccdc.cam.ac.uk).
Synthesis of the ligands and complexes
Synthesis of the ligands:
Synthesis of F-azosal
2-Fluoroaniline (3.72 g, 33.5 mmol) was dissolved in hydrochloric acid (6 M, 15 mL) and cooled to 3°C in an ice bath. Sodium nitrite (2.35 g, 34.8 mmol), dissolved in water (15 mL) was cooled below 5°C and added dropwise to the 2-fluoroaniline solution, maintaining a temperature below 5°C m/z (ESI, positive mode): 608.00 (35.33% [M+H]+). The resulting diazonium salt was coupled to salicylaldehyde by adding the salt to a cold solution of salicylaldehyde (3.51 mL, 33.5 mmol) dissolved in sodium hydroxide (10%, 15 mL), maintaining a temperature below 5°C. Cold water (50 mL) was added to the salicylaldehyde solution as a result the formation of a thick precipitate. Acetic acid (2 M, 15 mL) was added to the solution, and the solution was filtered using a Buchner funnel, washing with water. The crude product was recrytallised from CH3CN. Yield=3.31 g (40%). The purity was checked by elemental analysis. Calc. for C13H9FN2O2: C, 63.93; H, 3.71; N, 11.47. Found: C, 64.45; H, 3.67; N, 11.28. m/z (ESI, negative mode): 243.17 (100%, [M - H]-). FT IR (n/cm-1): 1664.11 (vs) (C=N). dH (400 MHz; CDCl3): 7.14-8.21 (aromatic H), 10.05 (s, OH), and 11.34 ppm (s, HC=O). dF (500 MHz; CDCl3): -108.5 ppm (tt).
Synthesis of sulf-azosal.0.75CH3CO2H.CH3CN
Sulfamerazine (8.85 g, 33.5 mmol) was dissolved in hydrochloric acid (6 M, 15 mL) and cooled to 3°C in an ice bath. Sodium nitrite (2.35 g, 34.8 mmol), dissolved in water (15 mL) was cooled below 5°C and added dropwise to the sulfamerazine solution, maintaining a temperature below 5°C. Cold water was added to the resulting diazonium salt solution, which was coupled to salicylaldehyde by adding the salt to a cold solution of salicylaldehyde (3.51 mL, 33.5 mmol) dissolved in sodium hydroxide (10%, 15 mL), maintaining a temperature below 5°C. Cold water (100 mL) was added to the salicylaldehyde solution as a result the formation of a thick precipitate. Acetic acid (2 M, 15 mL) was added to the solution, and the solution stirred for 25 minutes; then filtered using a Buchner funnel, washing with water. The crude solid product was orange in color. The product was recrystallized from hot acetonitrile. Yield=1.52 g (9%). The purity was checked by elemental analysis. Calc. for C21.5H21N6O5.5S: C, 53.41; H, 4.38; N, 17.38. Found: C, 53.14; H, 3.84; N, 17.45. m/z (ESI, negative mode): 396.00 (27.58% [M-H]-). FT IR (n/cm-1): 1657.15 (vs) (C=N). dH (500 MHz; CDCl3): 1.59 (s, N-H), 2.44 (s, 3H), 6.84-8.50 (aromatic H), and 10.07 ppm (s, HC=O).
Synthesis of the compounds:
Synthesis of compound 1
Compound 1 was synthesized and characterized as by Beltrán and co-workers [36]. m/z (ESI, positive mode): 485.92 (75.16% [M+H]+). dH (500 MHz; CDCl3): 6.72-8.75 ppm (aromatic H). dSn (500 MHz; CDCl3)=-327.42 ppm. Lit: [36] dSn (CDCl3)=-328.5 ppm. UV-Visible spectrum (DMSO), λmax./nm (10-4 e/M-1 cm-1): 268 (2.06), 386, sh (0.58), and 440 (1.19).
Synthesis of compound 2
F-azosal (0.56 g, 2.3 mmol) was refluxed in absolute ethanol (300 ml) for two minutes so as to ensure that all solids were dissolved; then 2-aminophenol (0.25 g, 2.3 mmol) and di-phenyltin oxide (0.66 g, 2.3 mmol) were added to the transcontrol homogeneous solution. The resulting mixture was refluxed for 18 hours during which time an orange precipitate was formed. The mixture was filtered hot; then the residue was washed with ethanol and air-dried. The filtrate was left to evaporate to leave crystals suitable for X-ray crystallogaphy. Total yield=1.07 g (77%). m/z (ESI, positive mode): 608.00 (35.33% [M+H]+). FT IR (n/cm-1): 1608.92 (vs) (C=N), 607.59 (m) (Sn-C), 537.89 (s) (Sn-O), and 445.84 (m) (Sn-N). dH (500 MHz; CDCl3): 6.74-8.88 ppm (aromatic H). dF (500 MHz; CDCl3): -109. 6 (tt) and -112.86 ppm (tt). dSn (500 MHz; DMSO-d6): -325.85 ppm. UV-visible spectrum (DMSO), λmax./nm (10-4 e/M-1 cm-1): 270 (5.32), 384 (1.69), and 442 (1.58).
Synthesis of compound 3
Sulfamerazineazosal.0.75CH3CO2H.CH3CN (0.22 g, 0.46 mmol) was refluxed in absolute ethanol (200 ml) for two minutes so as to ensure that all solids were dissolved; then 2-aminophenol (0.05 g, 0.46 mmol) and di-phenyltin oxide (0.13 g, 0.46 mmol) were added to the transcontrol homogeneous solution. The resulting mixture was refluxed for 24 hours during which time an orange precipitate was formed. The mixture was filtered hot; then the residue was air-dried. The orange product was recrystallised from CHCl3/hexane. Yield=0.24 g (67%). The purity was checked by elemental analysis. Calc. for C36.125H28.125Cl0.375N6O4SSn: C, 56.03; H, 3.66; N, 10.88. Found: C, 55.98; H, 3.69; N, 10.90. m/z (ESI, positive mode): 697.17 (100% [M-C5H6N3+H]+). FT IR (n/cm-1): 1605.11 (vs) (C=N), 614.64 (vs) (Sn-C), 520.05 (s) (Sn-O), and 446.65 (s) (Sn-N). dH (500 MHz; CDCl3): 2.44 (s, 3H) and 6.64-8.88 ppm (aromatic H). dSn (500 MHz; CDCl3): -325.77 ppm. UV-visible spectrum (DMSO), λmax./nm (10-4 e/M-1 cm-1): 262 (3.10), 301 (1.91), 396 (2.85), and 440 (2.80).
Cell culture
The human colon cell line HCT116 (American Type Culture Collection, Manassas, VA) was grown as monolayers in the medium suggested by American Type Culture Collection and supplemented with 10% fetal calf serum (Mediatech, Herndon, VA), penicillin (50 IU ml-1) and streptomycin (50 µl ml-1; Life Technologies, Grand Island, NY). Cells U-1242 MG cells were a kind gift from Isa Hussaini (University of Virginia). U-1242 MG cells were cultured in α-MEM media supplemented with 10% FBS (Invitrogen, Gaithersburg, MD) with the addition of penicillin (50 IU ml-1) and streptomycin (50 µg/ml; Life Technologies, Grand Island, NY) at 37°C in 5% CO2.
In vitro studies
Cell viability/growth was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay according to the manufacturer’s instructions (Roche Diagnostics, Nutley, NJ). In brief, cells were seeded in 96-well tissue culture plates at a density of 1 × 104 cells per well and allowed to adhere overnight. On the following day, cells were treated with compounds 1-3, or cisplatin at concentrations ranging from 1 to 1000 µM for up to 24 hours. At the designated time, 10 µl of MTT reagent I was added to the culture media and the plate returned to the 37°C incubator for 4 hours, at which time, 100 µl of the solubilization solution (MTT reagent II) was added and incubation continued overnight. Cellular proliferation was determined spectrophotometrically using an enzyme-linked immunosorbent assay (ELISA) plate reader at 590 nm. The MTT assay was validated in our system by the direct trypan blue method.
Cellular uptake
HCT 116 colon cancer cells were treated independently with each compound at a concentration of ½x, 1x, and 2x IC50 value for 24 hours. Cellular uptake was detected by spectral shifts (absorption 488 nm, emission: 615 nm) using FACScaliber (BD Bioscience) [40].
Cell cycle distribution
For cell cycle analysis, cells were stained with PI following standard protocols. Briefly, methanol-fixed control and treated cells were stained with 0.1 mg/ml propidium Iodide (PI)/100 μg ml-1 RNASE and fluorescence intensity was analyzed by FACScaliber (BD Bioscience). For each subset, we analyzed 10,000 events. All parameters were collected in listmode files. Data were analyzed using the software program WinMDI. The percentage of cells in G0-G1, G2-M, and S phases was determined from DNA content histograms.
Cell death
The annexin V assay was performed according to the manufacturer's instructions for “Rapid annexin V binding” (Oncogene Research Products, San Diego, CA). Briefly, cells post treatment were harvested, washed with ice-cold PBS, the media binding reagent annexin V-FTIC was added , and following a 15 minute incubation at room temperature, propidium iodide was also added. Cells were analysed immediately by flow cytometry. In addition, DNA fragmentation was assessed using the comet assay preformed as recommended by the manufacturer (Trevigen INC., Gaithersburg, MD). The extent of DNA fragmentation was determined using TriTek CometScore™ Freeware program.
Results and discussion
2,2-Diphenyl-6-aza-1,3-dioxa-2-stanna-[d,h]dibenzocyclononene (compound 1), compound 2, and compound 3 were synthesised from 1:1:1 stoichiometric amounts of the respective salicylaldehyde (salicylaldehyde, (E)-5-((4-fluorophenyl)diazenyl)-2-hydroxybenzaldehyde (F-azosal), or (E)-4-((3-formyl-4- Scheme 1. Synthesis of compounds 2 and 3.hydroxyphenyl)diazenyl)-N-(4-methylpyrimidin-2-yl)benzenesulfonamide.0.75CH3CO2H.CH3CN) (sulf-azosal.0.75CH3CO2H.CH3CN), 2-aminophenol, and di-phenyltin(IV) oxide in refluxing ethanol (Scheme 1). The synthesis of compound 1 has been previously reported by Beltrán and co-workers [36].
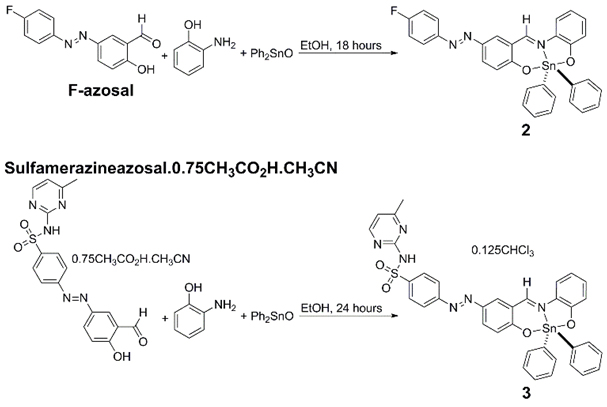
Scheme 1. Synthesis of compounds 2 and 3
All complexes were characterized by elemental analysis, ESI MS, UV-visible, fluorescence, 1H, 19F, and 119Sn, NMR spectroscopies, and X-ray crystallography, where appropriate.
ESI MS analysis
Low and high resolution ESI MS data were acquired for the ligands and compounds 1-3 (see ESI, Figures S1-S5). In all mass spectral analyses, we have assigned M as the molecular ion minus any solvates. In all of the ESI MS data, the respective species and their fragments were all detected in order to elucidate the respective structures. For 4-((E)-(4-fluorophenyl)diazenyl)-2-((E)-(2-hydroxyphenylimino)methyl)phenol (F-azosal), m/z (ESI, negative mode): 243.17 (100%, [M-H]-); 4-((E)-(4-hydroxy-3-((E)-(2-hydroxyphenylimino)methyl)phenyl)diazenyl)-N-(4-methylpyrimidin-2-yl) benzenesulfonamide.0.75CH3CO2H.CH3CN (sulf-azosal.0.75CH3CO2H.CH3CN), m/z (ESI, negative mode): 396.00 (27.58% [M-H]-); compound 1, m/z (ESI, positive mode): 485.92 (75.16% [M+H]+); compound 2, m/z (ESI, positive mode): 608.00 (35.33% [M+H]+); and compound 3, m/z (ESI, positive mode): 697.17 (100% [M-C5H6N3+H]+).
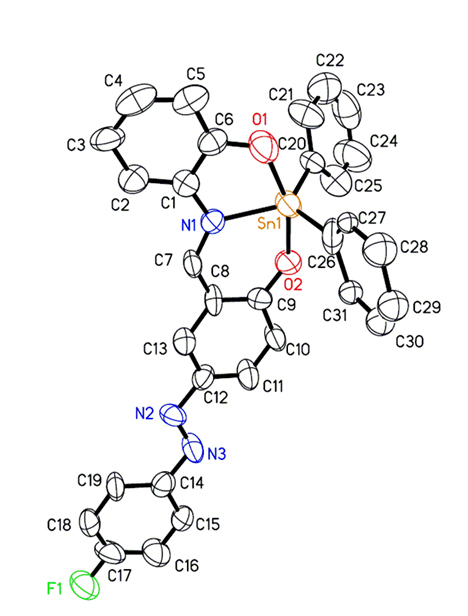
Figure 1. A thermal ellipsoid plot (50% probability envelopes) for compound 2.
X-ray crystallographic studies of compound 2
Crystals of compound 2 were grown by recrystallisation of the complex with ethanol. The structural elucidation of compound 2 was accomplished by X-ray crystallography. The molecular structure of compound 2, together with the atomic numbering scheme are shown in Figure 1; while Table 1 shows its crystal data and structure refinement (see ESI, Tables S1 and S2 for selected bond lengths and angles). The complex crystallized with trigonal bipyramidal geometry surrounding Sn(IV) metal centre. The O,N,O tridentate ligand places its two oxygen donating atoms in the axial positions, and the nitrogen atom occupies one equatorial position. The two phenyl groups attached to the Sn(IV) metal centre occupy the other two equatorial positions. The phenyl ring C26-C31 is disordered as two slightly offset rings with an equal distribution. Only one ring is included in the ORTEP representation as shown in Figure 1.
Compound reference |
shelxl |
Chemical formula |
C31H22FN3O2Sn |
Formula Mass |
606.21 |
Crystal system |
Orthorhombic |
a/Å |
10.207(3) |
b/Å |
11.209(3) |
c/Å |
45.443(11) |
α/° |
90.00 |
β/° |
90.00 |
γ/° |
90.00 |
Unit cell volume/Å3 |
5199(2) |
Temperature/K |
163(2) |
Space group |
Pbcn |
No. of formula units per unit cell, Z |
8 |
No. of reflections measured |
24333 |
No. of independent reflections |
4578 |
Rint |
0.1573 |
Final R1 values (I > 2σ(I)) |
0.1105 |
Final wR(F2) values (I>2σ(I)) |
0.2622 |
Final R1 values (all data) |
0.2040 |
Final wR(F2) values (all data) |
0.3411 |
Table 1. Crystal data and structure refinement for compound 2.
FT IR spectroscopic studies: IR spectra (see ESI, Figures S6-S10) of compounds 1-3 show two intense bands for the C=N fragment between 1607-1601 and 1568-1535 cm-1 [36,37]. These stretching frequencies are indirect evidences of the nature of the solid state structures as proven by X-ray crystallography for compound 2. Stretching frequencies for Sn–C, Sn–O and Sn-N bands were also observed, ranging from 604.75–614.64, 520.05–537.89, and 436.50-446.65 cm-1, respectively [36,37].
1H, 19F, and 119Sn NMR spectroscopic analysis
1H, 19F, and 119Sn NMR spectra were acquired for the respective ligands and complexes (see ESI, Figures S11-S19). 1H NMR spectroscopy was used to prove the structures of compounds 1-3 without any ambiguity. 19F NMR spectra were acquired for F-azosal and compound 2. Two chemical shifts were observed for F-azosal, with the major chemical shift occurring as triplets of triplets at -108.5 ppm for F-azosal, but two major chemical shifts were observed as triplet of triplets and septuplets for compound 2 at -109.6 and -112.9 ppm, respectively. This observation could be due to the existence of isomers in both compounds. In the near future, detailed 19F NMR spectroscopic studies will be carried out on compound 2 in an effort to fully ascertain the existence of the isomers (which can be either the trans or cis moiety).
119Sn NMR spectra were acquired for compounds 1-3 in the non-coordinating solvent, CDCl3, where the chemical shifts were -327.4, -325.9, and -325.8 ppm for compounds 1, 2, and 3, respectively. The chemical shift value of -328.5 ppm for compound 1 was reported in the literature [36]. These signals are indicative of a pentacoordinated Sn(IV) metal centre, which was also observed by Beltran and co-workers [36] and Sedaghat and co-workers [41] for analogous Sn(IV) complexes. The pentacoordinated Sn(IV) metal centre was also proven by X-ray crystallography for compound 2.
UV-visible and fluorescence spectroscopic studies of compound 1-3
UV-visible spectra were acquired for compounds 1-3 in DMSO (see ESI, Figure S20 and Table S3 for the various UV-visible spectra and spectroscopic data for compounds 1-3). Charge transfer bands in the region 262-270 nm, which can be assigned to π→π* transitions, were observed for compounds 1-3. This occurred at 268 nm for compound 1, 270 nm for compound 2, and 262 nm for compound 3, respectively. Charge transfer bands assigned to n→π* transitions of aromatic rings were observed in the region 301-396 nm. This was observed as a shoulder at 386 nm for compound 1, at 384 nm for compound 2, and at 301 and 396 nm for compound 3, respectively. In the UV range the tin(IV) complexes derived from salicylaldehyde generally possess a low-energy band in the range 384-396 nm which can be attributed to a π→π* transition originating mainly in the azomethine chromophore. This occurs as a shoulder at 386 nm for compound 1, as peaks at 384 and 396 nm for compound 2 and compound 3, respectively. A band around 440 nm was assigned to LMCT transitions of the type p→d where p and d represent phenolato oxygen’s lone pair and the tin(IV) metal centre for compounds 1-3. Fluorescence spectral data were acquired for compounds 1-3 in DMSO (see ESI, Figure S21). The excitation wavelength for compounds 1-3 was 440 nm. Compound 1, which contains no azo moiety, produced the highest intensity in its fluorescence spectra; while compounds 2 and 3 produced the lower intensities.
In vitro studies
The overall goal here was to synthesize chemopreventive agents that could potentially lead to their use in clinical evaluation. To this end, the anti-proliferative/apoptotic activity of the tin(IV) compounds described above were evaluated against human glioblastoma (U-1242 MG) and colorectal cancer cell lines (HT-29 and HCT-116).
Cell growth inhibition of U-1242 MG cells to azo-derived Sn(IV)-containing complexes
The rate of glioblastoma (GBM) cell proliferation was determined by measuring the amount of MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduced by U-1242 MG cells, following treatment with varying concentrations of three azo-derived compounds and a known inhibitor of GBM cells (LY 294002, PI3K inhibitor). Compound 1 reduced the relative rate of cell proliferation greater than 50% when used at 10 µM and 25% when used at 50 µM concentrations in comparison to cells stimulated with serum (10% FBS) and no inhibitor (Figure 2A). The compound 1-generated reduction in cell proliferation corresponds to the reduction of cell proliferation observed in cells treated with LY 294002 (50%) (Figure 2A). Since this compound demonstrated an ability to prevent GBM cell proliferation, the efficacies of two azo-containing tin complexes were next assayed for their ability to prevent GBM cell proliferation. Compound 2 demonstrated little anti-proliferative activity in the U-1242 MG cells and this reduction was only observed at the highest concentration (50 µM) (Figure 2B). Compound 3 showed an incremental reduction in the relative rate of cell proliferation as the concentration increased (Figure 2C). More importantly, compound 3 decreased cell proliferation at a higher rate than LY 294002 (Figure 2C). Taken together these data suggest that azo-containing Sn(IV) complexes are effective in preventing GBM cell proliferation.
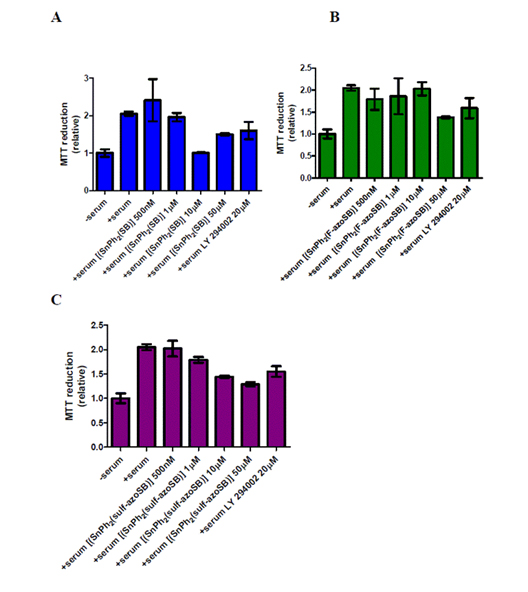
Figure 2. Effects of azo and non-azo containing tin complexes on glioblastoma cell proliferation. (A) [[Sn(Ph2SB)] (compound 1), (B) [[SnPh2(F-azoSB)] (compound 2), and (C) [[SnPh2(sulf-azoSB)] (compound 3) complexes were added to U-1242 MG cells that were cultured in serum (10% FBS) for 16 hours prior to experimentation. Cells were then exposed to each compound for 48 hours in the presence of serum. After this incubation, cells were treated with MTT, cultured for another 4.5 hours, reduced with a solution of SDS/HCL and MTT reduction measured via spectrophotometry. Each column represents the mean of three wells. Vertical bars indicate s.e.m. The columns of each graph are plotted as relative values in comparison to serum-starved cells (denoted as-serum on each graph).
Cell growth inhibition of four other cell lines to compounds 1-3 and cisplatin
We evaluated the effect of compounds 1-3 on the cancer cell lines (colorectal (HT-29, HCT-116) and skin (A431)) and a normal skin cell line (HFF). The evaluation involved the detection of alterations in cell cycle progression (PI staining), induction of necrosis/apoptosis (Annexin V-PI), DNA fragmentation (comet assays), and cell viability (MTT assay). Our objective was to compare and contrast the effectiveness of compounds 1-3 with the control compound, cisplatin. Cell viability/growth was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay as described in the experimental section. As shown in Table 2, after 24 hours of treatment, IC50 values for compounds 1-3 ranged from 3.04 ± 0.98 to 104.51 ± 13.87 mM. Compounds 1-3 decreased cellular growth in all cell lines tested; however, they were more effective against the A431 skin and HCT 116 colon cancer cells. Under the same conditions, a decrease in cellular growth of all cell lines tested was considerably lower than for cisplatin; the IC50 values for cells treated with cisplatin were determined to range between 29.73 ± 8.70 to 269.96 ± 93.90 mM.
|
Compounds and IC50 values (µM) |
Cell line |
1 |
2 |
3 |
4 |
Colon |
|
|
|
|
HT-29 |
5.01 ± 0.44 |
104.51 ± 13.87 |
7.52 ± 0.94 |
151.37 ± 7.30 |
HCT116 |
3.7 ± 0.65 |
89.45 ± 18.07 |
9.03 ± 3.32 |
269.96 ± 93.90 |
Skin |
|
|
|
|
HFF |
16.45 ± 0.94 |
>1000 |
19.1 ± 4.50 |
91 ± 2 |
A431 |
3.04 ± 0.98 |
103.64 ± 19.03 |
3.28 ± 0.92 |
29.73 ± 8.70 |
Table 2. Growth inhibition of human cancer cell lines by the compounds. Compounds 1 ([[Sn(Ph2SB)]), 2 ([[SnPh2(F-azoSB)]), 3 ([[SnPh2(sulf-azoSB)]), and 4 (cisplatin)
Interestingly, cisplatin was the least effective of the four compounds tested against HCT 116 colon cancer cells. The IC50 value for cisplatin was seventy-two times higher than that of compound 1. Overall, compounds 2 and 3 were much more effective compared to the control compound cisplatin. In all subsequent studies, cells were treated at a concentration of ½ x, 1 x, and 2 x IC50 value.
Uptake of compounds 1-3 was detected via flow cytometry
Based on fluorescence shifts, we were able to discern cellular uptake of compounds 1-3 into HCT116 colon cancer cells. Figure 3 shows that compound 2 was readily taken up by the cells at each of the three concentrations examined: whereas, compound 1 had a marginal shift at ½x and 1x IC50 value. However, at an IC50 value of 2x, the fluorescence shift was somewhat comparable to compound 2 at its 2x IC50 value. For compound 3, the shift at all concentrations was less than those seen for compounds 1 and 2. However, in all cases the fluorescence shift (i.e., sample uptake) was concentration dependent.
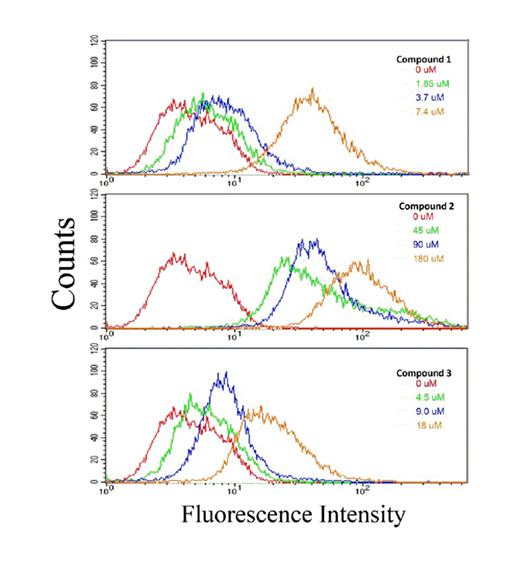
Figure 3. Cellular uptake of compounds 1, 2, and 3 by HCT 116 colon cancer cells. HCT 116 cells were treated at the designated concentration for 24 hours with compounds 1, 2, or 3. Uptake of compounds was determined by flow cytometric analysis; described in the Materials and Methods section. A fluorescence spectral shift is an indication of cellular uptake of drug. The data presented here is representative of two experiments in which results were comparable.
Compounds 1-3 arrested cell cycle transition and induced apoptosis/necrosis
It was found that compounds 1-3 interrupted cell cycle progression and induced cell death. Cellular response of HCT 116 colon cancer cells to cisplatin and compounds 1-3 was evaluated using several methods of detection (described in experimental section). In doing so, we were able to analyse the effect of each compound on cell cycle transition and cell death. As seen in Figure 4A (top), compound 1 blocked cell cycle transition at the G2-M phase. This is evident by a decrease in the number of cells in G1 and S phases with a concomitant increase in G2. Compound 1 also induced cell death as demonstrated in Figure 4A (middle). We conclude that the mechanism of action of compound 1 may in part be due to its effect on nuclear DNA, as seen by the fragmentation of DNA in Figure 4A (bottom). It is evident by visual inspection that this effect is concentration-dependent and is seen at a concentration lower than the IC50 value for growth.
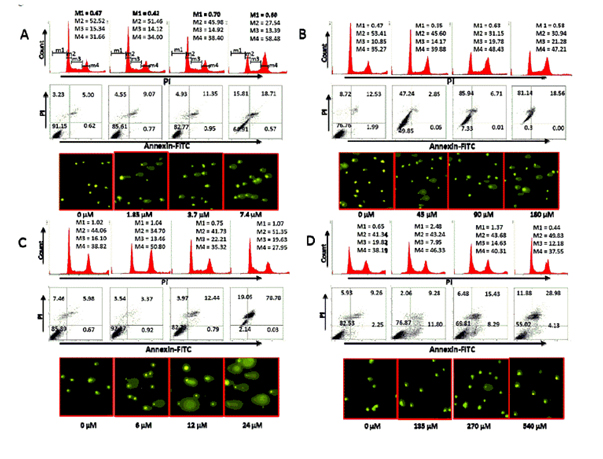
Figure 4. Effect of compounds 1 (A), 2 (B), 3 (C), and 4 (D) on cell cycle progression and cell death in HCT 116 colon cancer cells. Cells were treated independently with each compound at the designated concentration for 24 hours. After treatment, the cells were analysed, as described in the Materials and Methods section, for cell cycle transition (Top), cell death (middle) and DNA fragmentation (bottom). M1, M2, M3 and M4 correspond to sub-G0-G1, G0-G1, S and G2-M phases, respectively. All data are from a single experiment. This study has been repeated three times generating results within 10% of those presented here.
Similar effects were noted for compounds 2 and 3 (Figure 4B and 4C). However, the extent of cell death was greatest with compound 2 (Figure 4B middle). Here, we noted that more than 50% of the cells treated with compound 2 were annexinV+ and/or PI+ at ½ x IC50 value. Interestingly, of all the samples examined, DNA fragmentation was more pronounced in cells treated with compound 3. The qualitative extent of DNA damage in the cells treated with all samples was measured by the length of DNA migration and the percentage of migrated DNA using the TriTek CometScore™ freeware program. The overall percent of DNA in the tail of the comet, as an indication of DNA fragmentation, is a direct correlation to concentration. As shown in Table 3, DNA fragmentation in cells treated with compound 3 was more pronounced than with the three other compounds examined. The lowest percentage of fragmentation was seen in cells treated with the control compound cisplatin (compound 4).
|
|
Compound |
µM |
1 |
2 |
3 |
4 |
0 |
11.6 ± 2.9 |
11.1 ± 2.2 |
16.0 ± 4.7 |
6.7 ± 1.2 |
½ x IC50 |
34.1 ± 6.6 |
20.4 ± 8.1 |
7.3 ± 3.8 |
IC50 |
24.8 ± 3.6 |
14.8 ± 3.8 |
36.6 ± 7.5 |
11.2 ± 3.4 |
2 x IC50 |
43.2 ± 5.3 |
26.0 ± 4.0 |
71.4 ± 7.1 |
14.6 ± 3.8 |
Table 3. Comet assay of HCT 116 cancer cells treated with compounds 1, 2, 3, or 4.
Numbers are expressed as means ± SE
Units expressed as overall percent of DNA in the tail of the comet
Again, the cellular effect of the control compound cisplatin was reduced compared to compounds 2 and 3. Cisplatin had a marginal effect on cell cycle transition (Figure 4D top). At the highest concentration examined (2 x IC50 value) only about 25% of the cells were necrotic or apoptotic (Figure 4D middle). Similarly, DNA fragmentation (Figure 4D bottom; Table 3) was least pronounced in HCT 116 cells treated with cisplatin. Therefore, we conclude that compounds 2 and 3 are more effective than cisplatin. Similar effects were observed when HT-29, A431, and HFF cell lines were treated with these compounds (data not shown). Here, Figure 4 is representative of the results obtained with all four cell lines.
Many studies have reported inhibition of proliferation and induction of apoptosis in response to various anti-cancer reagents like NO-aspirin [42,43], quercetin [44-46], curcumin [47-49], and resveratrol [50,51]. Here, we report that compounds 1-3 induced colon cancer cell death via an anti-proliferative/proapoptotic mechanism. This effect was observed in several cell lines; thus, demonstrating a tissue-independent effect. In our studies, the IC50 values of compounds 1-3 for the inhibition of cell growth varied within a relatively narrow range, depending on the cell line. However, in all instances, it has been remarkably low, being at the most seventy-two times lower than that of the control compound, cisplatin. On the whole, our data show a higher efficacy with compounds 1-3 than in the control compound, cisplatin.
Although it is difficult to assess the contribution of these changes to the final cell number, it appears that both the anti-proliferative and proapoptotic effects of the cisplatin derivative compounds make an important contribution to its cell growth inhibitory effect, with perhaps the cell death effect predominating. The overall contribution of compounds 1-3 to blocking the cell cycle is difficult to quantify.
Taken together these data suggests the chemical composition of compounds 1 and 3 would serve as effective anti-proliferative agents. We can interpret from these data that the various tin compounds share a property that affects processes common to all cancer cell lines (i.e., increased proliferation and aberrant apoptosis). Several researchers have reported that organotin(IV) complexes have the ability to induce apoptotic cell death [52]. However, the actual mechanism of this induced apoptosis of organotin(IV) compounds has not yet been determined. Today, it is believed that organotin(IV) compounds have the ability to react with cell membranes while leading to their decay, enhancing the ion exchange process, and finally inhibiting the oxidative and photochemical phosphorylation [53].
It is known that organotin(IV) compounds could be involved in other biological processes that occur in cells, for example, in peroxide oxidation of lipids [53]. Now, acceleration of peroxide oxidation of lipids in cells can lead to accumulation of hydroperoxides, decay of cell membranes, and various pathologies in living cells [53]. As it is known that organotin(IV) complexes can exhibit redox properties [54], it was assumed that their toxicity originates from interaction with reductants in biomolecules. Reactions of organotin(IV) complexes with phosphorus-containing biomolecules, such as phospholipids, ATP, and nucleic acids, were shown to inhibit the synthesis of phospholipids and their intracellular transport [53,54], which may be responsible for the anti-proliferative activity of organotin(IV) derivatives, inclusive of compounds 1-3 that are reported in this study.
Also, as reported by Pellerito et al. [53], triorganotin(IV) complexes appear to inhibit the mitochondrial function in at least three ways: (i) by causing large-scale swelling at high concentrations, (ii) mediating Cl-/OH- exchange across membranes, and (iii) inhibiting oxidative phosphorylation or ATP hydrolysis. It was also inferred that the organotin(IV) mediated anion exchange across the mitochondrial membrane may also interfere with ATP synthesis or hydrolysis. These modes of action can possibly account for the cytotoxic effects as exhibited by our tin(IV) complexes (compounds 1-3).
Conclusions
Novel ligands and tin(IV) compounds 2, and 3, along with the reported compound 1, were successfully synthesized and characterised. In summary, the anti-cancer properties of compounds 1-3 were evaluated in two colon cancer cell lines, HT-29 (data not shown) and HTC-116. In all instances, results for the two cell lines were consistent despite genetic variability within the cell lines. Our results show a progressive uptake of drug (concentration dependent) and, in direct response, we have demonstrated that each compound inhibited proliferation and induced apoptosis and nuclear fragmentation in a similar fashion.
In conclusion, our findings document that compounds 1-3 have the potential to induce an array of potentially important molecular changes in the malignant colonocyte that may be relevant to its pharmacological actions in cancer. Discovering compounds which exhibit anti-proliferative activity on cancer cells, but do not affect non-cancerous cells, is the characteristic objective of any chemotherapy/chemopreventive research. Thus, our overall aim is to formulate chemopreventive agents that target with specificity only cancer cells. An encouraging aspect of this type of research is that data generated from multiple ongoing intervention trials could be used to define in cancer cells the cancer preventive activity of novel chemical compounds and supply us with information that could lead to the formulation of more effective and less toxic chemopreventive agents. Thus, the assimilation of newly available pharmacokinetic information will assist us in improving upon now available chemopreventive agents.
In this regard, the following are important follow up studies: (1) determination of structural relationships) and (2) differentiation of downstream mediators of the apoptotic and cell cycle effects to assess which pathways the derivatives of cisplatin recruit to augment the effect of the control compound (cisplatin). Here we propose the potential usefulness of cisplatin derivatives for cancer treatment or prevention. Our next step is to extrapolate our cell culture findings and eventually evaluate their relevance in chemoprevention of human colon cancer by testing in animal tumour models; the ultimate goal being a transition study that would include humans.
Acknowledgements
This work was supported in part by the Mississippi INBRE funded by grants from the National Centre for Research Resources (5P20RR016476-11) and the National Institute of General Medical Sciences (8 P20 GM103476-11) from the National Institutes of Health; and KO1 CA106604 (JLW) and CA140487 (JLW) funded by the National Cancer Institute. The authors also acknowledge the NSF for funding the ESI and MALDI-ToF mass spectrometers (Grant # CHE 0639208).
We are also grateful for the use a 400 MHZ NMR spetrometer, which was funded by the NSF CRIF:MU Award # 0840390. AAH is grateful to Old Dominion University for the start up package which allowed for the completion of this research project. AAH is also grateful for the USM Lucas Endowment Grant and funding from ExxonMobil Research and Engineering Company through Dr. John Robbins. AAH would like to thank Mr. Joshua D. Phillips for acquiring the ESI MS, UV-visible, and fluorescence data for this manuscript. We would like to thank Dr. Vijayaraghavan Rangachari and his research group for the use of their Cary Eclipse fluorescence spectrophotometer. We would also like to thank Professor Glen Shearer for his contant positive motivation which helped mobilize this project. AAH would also like to thank Old Dominion University’s Faculty Proposal Preparation Program (FP3) and also for the Old Dominion University start-up package that allowed for the successful completion of this work. The authors are also very grateful for the helpful comments that were provided by the reviewers.
References
- Vande Woude GF, Klein G (2000) Advances in Cancer Research. Academic Press San Diego, California, USA 87: 1-252.
- Cardin CJ, Roy A (1986) Anticancer activity of organometallic compounds. The reaction of dimethyltin dichloride with nucleosides under biologically relevant conditions. Inorg Chim Acta 125: 63-66.
- Pellerito L, Nagy L (2002) Organotin(IV)n+ complexes formed with biologically active ligands: equilibrium and structural studies, and some biological aspects. Coord Chem Rev 224: 111-150.
- Yang P, Guo M (1999) Interactions of organometallic anticancer agents with nucleotides and DNA. Coord Chem Rev 185: 189-211.
- Wang X, Zhang X, Lin J, Chen J, Xu Q, et al. (2003) DNA-binding property and antitumor activity of bismuth(III) complex with 1,4,7,10-tetrakis(2-pyridylmethyl)-1,4,7,10-tetraazacyclododecane. Dalton Trans: 2379-2380.
- Chakraborty S, Bera AK, Bhattacharya S, Ghosh S, Pal AK, et al. (2002) Synthesis, microbiological study and X-ray structural characterisation of a tri-n-butylstannyl-2[4(diethylamino)arylazo]benzenecarboxylate. J Organomet Chem 645: 33-38.
- Kidwai M, Dave B, Misra P, Saxena RK, Singh M (2000) Novel synthetic approach for antifungal and antibacterial organotin compounds. Inorg Chem Commun 3: 465-468.
- Sharma V, Sharma RK, Bohra R, Ratnani R, Jain VK, et al. (2002) Synthesis, spectroscopic and structural aspects of some tetraorganodistannoxanes with internally functionalized oxime: crystal and molecular structures of [{R2Sn(ONC(Me)py)}2O]2 (R=Bun and Et) and 2-NC5H4C(Me)NOHJ. J Organomet Chem 651: 98-104.
- Tabassum S, Pettinari C (2006) Chemical and biotechnological developments in organotin cancer chemotherapy. J Organomet Chem 691: 1761-1766.
- Arjmand F, Yousuf I, Zaidi Y, Toupet L (2015) Crystal structure determination, spectroscopic characterization and biological profile of a tailored ionic molecular entity, Sn(IV) iminodiacetic acid–piperazinediium conjugate: in vitro DNA/RNA binding studies, Topo I inhibition activity, cytotoxic and systemic toxicity studies. RSC Adv 5: 16250-16264.
- Zaidi Y, Arjmand F, Zaidi N, Usmani JA, Zubair H, et al. (2014) A comprehensive biological insight of trinuclear copper(II)-tin(IV) chemotherapeutic anticancer drug entity: in vitro cytotoxicity and in vivo systemic toxicity studies. Metallomics 6: 1469-1479. [Crossref]
- Navakoski de Oliveira K, Andermark V, von Grafenstein S, Onambele LA, Dahl G, et al. (2013) Butyltin(IV) benzoates: inhibition of thioredoxin reductase, tumor cell growth inhibition, and interactions with proteins. Chem Med Chem 8: 256-264. [Crossref]
- Duanmu J, Cheng J, Xu J, Booth CJ, Hu Z (2011) Effective treatment of chemoresistant breast cancer in vitro and in vivo by a factor VII-targeted photodynamic therapy. Br J Cancer 104: 1401-1409. [Crossref]
- Cima F, Ballarin L (1999) TBT-induced apoptosis in tunicate haemocytes. Appl Organomet Chem 13: 697-703.
- Pellerito C, D'Agati P, Fiore T, Mansueto C, Mansueto V, et al. (2005) Synthesis, structural investigations on organotin(IV) chlorin-e6 complexes, their effect on sea urchin embryonic development and induced apoptosis. J Inorg Biochem 99: 1294-1305. [Crossref]
- Xiao J, Cui J, Su Y, He J, Yao J (1993) J Chinese Pharm Sci 2: 45-52
- Blower PJ (2004) 30 Inorganic pharmaceutical. Annu Rep Prog Chem Sect A 100: 633-658.
- Chasapis CT, Hadjikakou SK, Garoufis A, Hadjiliads N, Bakas T, et al. (2004) Organotin(IV) derivatives of L-cysteine and their in vitro anti-tumor properties. Bioinorg Chem Appl. [Crossref]
- Höti N, Ma J, Tabassum S, Wang Y, Wu M (2003) Triphenyl tin benzimidazolethiol, a novel antitumor agent, induces mitochondrial-mediated apoptosis in human cervical cancer cells via suppression of HPV-18 encoded E6. J Biochem 134: 521-528. [Crossref]
- Höti N, Zhu DE, Song Z, Wu Z, Tabassum S, et al. (2004) p53-dependent apoptotic mechanism of a new designer bimetallic compound tri-phenyl tin benzimidazolethiol copper chloride (TPT-CuCl2): in vivo studies in Wistar rats as well as in vitro studies in human cervical cancer cells. J Pharmacol Exp Ther 311: 22-33. [Crossref]
- Chen F, Vallyathan V, Castranova V, Shi X (2001) Cell apoptosis induced by carcinogenic metals. Mol Cell Biochem 222: 183-188. [Crossref]
- Gennari A, Bleumink R, Viviani B, Galli CL, Marinovich M, et al. (2002) Identification by DNA macroarray of nur77 as a gene induced by di-n-butyltin dichloride: its role in organotin-induced apoptosis. Toxicol Appl Pharmacol 181: 27-31. [Crossref]
- Boualam M, Biesemans M, Meunier-Piret J, Willem R, Gielen M (1992) In vitro antitumour activities of a novel 2:3 condensation product of salicylaldoxime with di-n-butyltin(IV) oxide. Appl Organomet Chem 6: 197-205.
- Gielen M, Biesemans M, Willem R, Tiekink ERT (2004) Diorganotin Salicylaldoximate Clusters: Their Fascinating Chemistry and Structures. Eur J Inorg Chem: 445-451.
- Saxena A, Tandon JP (1983) Antitumor activity of some diorganotin and tin(IV) complexes of Schiff bases. Cancer Lett 19: 73-76. [Crossref]
- Saxena A, Tandon JP, Crowe AJ (1985) Synthesis and spectroscopic studies on organotin derivatives of biologically active Schiff bases. Polyhedron 4: 1085-1089.
- Tian L, Qian B, Sun Y, Zheng X, Yang M, et al. (2005) Synthesis, structural characterization and cytotoxic activity of diorganotin(IV) complexes of N-(5-halosalicylidene)-α-amino acid. Appl Organomet Chem 19: 980-987.
- Tian L, Sun Y, Qian B, Yang G, Yu Y.et al. (2005) Synthesis, characterization and biological activity of a novel binuclear organotin complex, Ph3Sn(HL)·Ph2SnL [L = 3,5-Br2-2-OC6H2CH[DOUBLE BOND]NCH(i-Pr)COO]. Appl Organomet Chem 19: 1127-1131.
- Gómez E, Contreras-Ordoñez G, Ramírez-Apan T (2006) Synthesis, characterization and in vitro cytotoxicity of pentacoordinated Tin(IV) complexes derived from aminoalcohols. Chem Pharm Bull (Tokyo) 54: 54-57. [Crossref]
- Langner M, Gabrielska J, Przestalski S (2000) Adsorption of phenyltin compounds onto phosphatidylcholine / cholesterol bilayers. Appl Organomet Chem 14: 25-33.
- Miszta A, Gabrielska J, Przestalski S, Langner M (2005) The effect of phenyltin chlorides on osmotically induced erythrocyte haemolysis. Appl Organomet Chem 19: 736-741.
- Olzynska A, Przybylo M, Gabrielska J, Trela Z, Przestalski S, et al. (2005) Di- and tri-phenyltin chlorides transfer across a model lipid bilayer. Appl Organomet Chem 19: 1073-1078.
- Rozycka-Roszak B, Pruchnik H, Kaminski E (2000) The effect of some phenyltin compounds on the thermotropic phase behaviour and the structure of model membranes. Appl Organomet Chem 14: 465-472.
- Muhammad N, Zia ur R, Ali S, Meetsma A, Shaheen F (2009) Organotin(IV) 4-methoxyphenylethanoates: Synthesis, spectroscopic characterization, X-ray structures and in vitro anticancer activity against human prostate cell lines (PC-3). Inorg Chim Acta 362: 2842-2848.
- Wiecek J, Dokorou V, Ciunik Z, Kovala-Demertzi D (2009) Diorganotin Complexes of a Thiosemicarbazone, Synthesis: Properties, X-Ray Crystal Structure, and Antiproliferative Activity of Diorganotin Complexes. Polyhedron 28: 3298.
- Beltran HI, Damian-Zea C, Hernandez-Ortega S, Nieto-Camacho A, Ramirez-Apan MT (2007) Synthesis and characterization of di-phenyl-tinIV-salicyliden-ortho-aminophenols: Analysis of in vitro antitumor/antioxidant activities and molecular structures. J Inorg Biochem 101: 1070-1085.
- Molecular Structure Corporation & Rigaku (2006) CrystalClear 14 MSC, The Woodlands, Texas, USA, and Rigaku Corporation, Tokyo, Japan
- Jacobson RA (1998) REQAB Version 11 In, The Woodlands, Texas, USA
- Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr Sect A: Found Crystallogr A64: 112-122.
- Gottschaldt M, Schubert US, Rau S, Yano S, Vos JG, et al. (2010) Sugar-Selective Enrichment of a D-Glucose-Substituted Ruthenium Bipyridyl Complex Inside HepG2 Can. Chem Bio Chem 11: 649-652.
- Sedaghat T, Shafahi F (2009) Synthesis, spectral, and thermal studies of organotin(IV) complexes with 4-bromo-2-{[(2-hydroxyphenyl) imino]methyl}phenol. Main Group Chem 8: 1-9.
- Williams JL, Borgo S, Hasan I, Castillo E, Traganos F, et al. (2001) Nitric Oxide-releasing Nonsteroidal Anti-inflammatory Drugs (NSAIDs) Alter the Kinetics of Human Colon Cancer Cell Lines More Effectively than Traditional NSAIDs: Implications for Colon Cancer Chemoprevention. Cancer Research 61, 3285-3289.
- Konturek PC, Kania J, Burnat G, Hahn EG (2006) NO-releasing aspirin exerts stronger growth inhibitory effect on Barrett's adenocarcinoma cells than traditional aspirin. J Physiol Pharmacol 57 Suppl 12: 15-24. [Crossref]
- Liang W, Li X, Li C, Liao L, Gao B, et al. (2011) Quercetin-mediated apoptosis via activation of the mitochondrialdependent pathway in MG-63 osteosarcoma cells. Mol Med Report 4: 1017-1023.
- Gibellini L, Pinti M, Nasi M, Montagna Jonas P, De Biasi S, et al. (2011) Quercetin and cancer chemoprevention. Evid Based Complement Alternat Med: 591356. [Crossref]
- Senthilkumar K, Arunkumar R, Elumalai P, Sharmila G, Gunadharini DN, et al. (2011) Quercetin inhibits invasion, migration and signalling molecules involved in cell survival and proliferation of prostate cancer cell line (PC-3). Cell Biochem Funct 29: 87-95. [Crossref]
- Fossey SL, Bear MD, Lin J, Li C, Schwartz EB, et al. (2011) The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 11: 112. [Crossref]
- Song MY, Yim JY, Yim JM, Kang IJ, Rho HW, et al. (2011) Use of curcumin to decrease nitric oxide production during the induction of antitumor responses by IL-2. J Immunother 34: 149-164. [Crossref]
- Liao S, Xia J, Chen Z, Zhang S, Ahmad A, et al. (2011) Inhibitory effect of curcumin on oral carcinoma CAL-27 cells via suppression of Notch-1 and NF-κB signaling pathways. J Cell Biochem 112: 1055-1065. [Crossref]
- Araújo JR, Gonçalves P, Martel F (2011) Chemopreventive effect of dietary polyphenols in colorectal cancer cell lines. Nutr Res 31: 77-87. [Crossref]
- Yang R, Zhang H, Zhu L (2011) Inhibitory effect of resveratrol on the expression of the VEGF gene and proliferation in renal cancer cells. Mol Med Rep 4: 981-983. [Crossref]
- Assunta Girasolo M, Rubino S, Portanova P, Calvaruso G, Ruisi G, et al. (2010) New organotin(IV) complexes with l-Arginine, N α - t-Boc- l-Arginine and l-Alanyl- l-Arginine: Synthesis, structural investigations and cytotoxic activity. J Organomet Chem 695: 609-618.
- Pellerito C, Nagy L, Pellerito L, Szorcsik A (2006) Biological activity studies on organotin(IV)n+ complexes and parent compounds. J Organomet Chem 691: 1733-1747.
- Das VGK (1996) Main Group Eelements and Their Compounds, Springer, New Delhi, India, ISBN: 9783540614258: 422.