Blood collection protocols are a crucial pre-analytical variable that must be carefully defined during the development of any reproducible biomarker assay. It is essential to limit endogenous proteolysis during collection and preservation of the plasma sample in a state compatible with down-stream analysis. This study tested the proteome and peptidome stability of matched plasma samples collected from healthy individuals. Each set of samples was donated simultaneously and stored/handled identically using three different types of blood collection tubes; (i) spray-dried EDTA (ii) EDTA with polymer gel, and (iii) EDTA tubes containing a proprietary cocktail of protease inhibitors (P100TM). All samples were investigated with proteomic and peptidomic approaches using 2D difference gel electrophoresis (2D DIGE) and solid phase extraction (SPE) followed by ESI-MS/MS (Electrospray Ionization) analysis. Results from 2D DIGE showed no significant influence of the different anticoagulant tubes on the level of plasma proteins observed by 2D DIGE, although minor variations in one protein per individual in each individual were observed, which statistically appears to be a random occurrence. In contrast, results obtained from the peptidomic analysis demonstrated a lower number of endogenous peptides suggesting preservation of some proteins in plasma treated with the protease inhibitor cocktail. However every plasma sample analysed showed endogenous peptides obtained from six proteins (namely Fibrinogen alpha chain, Complement C4-A, Complement C4-B, Alpha-2-antiplasmin, Complement C3 and Apolipoprotein E) irrespective of the anticoagulant used for collection. These data should assist researchers in their choice of ideal anticoagulants for plasma proteome and peptidome research.
Clinical proteomics aims to discover, verify and validate disease-specific protein biomarkers that can be applied for diagnostic, prognostic or therapeutic purposes [1-4]. Blood is the most commonly available biological sample for proteomic analysis leading to the identification of biomarkers [5]. Biomarkers can exist in a variety of different molecular and cellular forms e.g., proteins, DNA, circulating tumor cells. Development of biomarker assays and analytical validation are complex processes, that need to consider a number of potential factors (analytical variables, biological variables and cohort composition) and the nature of the application (e.g., invasiveness of the test, privacy, patient compliance and cost) [6]. In order to identify and quantify a biomarker accurately, and to ensure the clinical success of candidate biomarkers, they must be stable during sample collection, preparation and purification and must be present at a detectable concentration. Therefore the preanalytical and storage conditions [7,8] need to be carefully established to prevent any degradation during sample handling, processing, transfer or storage [9]. Plasma profiling is an essential part of proteomic research that combines protein separation and characterization technologies [10-12]. Following individual attempts by several industry and clinical proteomics/peptidomics research groups, the HUPO Plasma Proteome Project [13-15] was established in 2002 with the goal of comprehensively characterising the protein constituents of human plasma. The project involved 35 laboratories from around the globe and initially identified 3,020 plasma proteins [16] with high stringency (two or more peptides). Most laboratories focussed exclusively on plasma proteins and contributed to one of the major aims, namely that of creating a plasma proteome inventory. Although there was a growing awareness that reproducibility would be compromised if variability between samples was ignored, the amount of data required to substantiate the effect of such variables on results obtained from proteomics experiments from plasma was not insignificant [17]. In 2005, the HUPO Plasma Proteome Project (PPP) [17] Sample Collection and Handling Committee released a list of variables that need to be considered for the plasma collection and handling process (sample type, collection system, handling issues e.g., stabilization, processing storage and potential effects of additives) [18,19]. A number of plasma proteomic studies have reported that many of the observed peptides were generated through the activity of endogenous proteases [20] (e.g. amino peptidases [21] and carboxypeptidases [22]). Proteolysis commonly occurs as a time dependant degradation process that can significantly influence comparative analyses between blood samples [20,23,24]. Therefore complications and lack of consistency in the sample collection process can hinder the discovery and accurate measurement of protein biomarkers from plasma [25]. It is clear therefore, that a standardized blood sample collection strategy is essential to limit the generation of ex vivo artefacts [26] which not only involves a suitable collection process but also the preservation of the sample in a stable state suitable for downstream analysis [27]. For this reason the idea of blood collection tubes containing protease inhibitors was introduced to control sample degradation by minimizing proteolysis [28]. However, additional aspects of the blood collection protocol (i.e. use of anticoagulant in collection tube, centrifugation speed, centrifugation time) have met the attention of the research community [29-31] and will also play an essential role in obtaining quality data for downstream data processing [32].
Common additives in plasma preparation methodologies include anticoagulants such as ethylenediaminetetraacetic acid (EDTA), sodium citrate, heparin and warfarin [28]. However citrate, heparin and warfarin are not suitable for downstream proteomic analysis for various reasons [33-35]. Citrate, as a liquid reagent contained in the blood-drawing tube, generally dilutes the plasma sample which needs to be taken into account during any further study [36]. In addition, citrate can bind calcium and has resulted in falsely lowered readings of immunoassay measurements for multiple analytes [19]. Heparin is a polymeric glucosaminoglycan, which shows up as a characteristic repeating building-block polymer in MS, masking signals from protein and peptide components and rendering downstream MS analysis challenging [36]. A more commonly used and MS compatible additive is EDTA, a polyprotic acid containing four carboxylic acid groups and two amine groups with lone-pair electrons, that chelates calcium and several other metal ions[37,38]. Although EDTA inhibits the coagulation pathway and prevents clot formation, it does not prevent the activation of the first enzymatic steps for which the possibility of consequent ex-vivo generation of peptides still remains [38]. To avoid protease activity during and after blood collection, and to address the disadvantages mentioned above, tubes containing protease inhibitors (PI) have been designed which are intended to stabilize the blood proteome during the first 15 min of blood extraction. To date, to our knowledge, no comprehensive study has been performed to investigate the efficacy of the different blood collection tubes available in terms of low abundant proteins or at the endogenous/non-tryptic peptide level.
Our current study was designed to assist proteomic researchers by establishing a cost effective and standardized collection protocol that will preserve the plasma samples at an optimum condition for downstream analysis. There are currently three major varieties of EDTA containing tubes that are commercially available that use: EDTA only as anticoagulant (‘E’: BD Vacutainers EDTA Tube), a related EDTA containing tube has a dense polymer gel base layer which, upon centrifugation, partitions cellular bodies into the gel (‘G’: BD Vacutainers Plasma Preparation Tube PPTTM) and an EDTA containing tube with a proprietary cocktail of protease inhibitors (PIs) (‘P’: BD P100TM Blood Collection and Preservation System for Plasma Protein Analysis) [39]. The dense gel in ‘G’ tube forms a physical barrier between plasma and blood cells during centrifugation. The ‘P’ tube claims to reduce sample degradation by endogenous proteases. However, it needs to be attached to a specially designed blood collection kit to prevent backflow of the blood and avoid adverse patient reaction from contamination with protease inhibitors. Furthermore, a mechanical separator is required that will provide a solid barrier between plasma and cellular material during centrifugation. The P tubes with the complete blood collection kit costs US$30.00 while the EDTA containing tubes (E tube and G tube) are far cheaper (US$0.04-0.08 and US$0.04-0.1 respectively). It has been claimed that the P tubes are able to stabilize proteins and minimize proteolytic degradation during the collection and processing of blood specimens. Several studies have evaluated the blood collected in the P tubes against conventional collection tubes using targeted proteomic approaches e.g., selected/multiple reaction monitoring (SRM or MRM)- MS [26,40]. Our study was designed to estimate the intrinsic protease activity in plasma samples collected in the three commonly used tubes- EDTA, Gel EDTA and P100 by the differential comparison of low abundance proteins and endogenous peptides using 2D DIGE (2 Dimensional Differential Gel Electrophoresis) and solid phase extraction (SPE). The 2D DIGE technique which is based upon fluorescence prelabeling of protein mixtures before 2D gel electrophoresis, has been adopted in this study specifically to investigate low abundance proteins [41]. This technique combines the benefits of protein separation on a 2D gel as well as visualization of two different samples on a single gel [42]. Our previous studies have shown that commercial SPE cartridges offer the most reproducible peptide recoveries from EDTA plasma in technical replicates [43]. Therefore we have adopted the SPE technique to detect differences in the endogenous peptides between three different plasma preparations. This study will evaluate the proteomic and peptidomic profiles of the plasma samples and evaluate the efficacy of protease inhibitor containing EDTA tubes against standard EDTA tubes for downstream proteomic and peptidomic analysis.
Blood (10 ml) was collected from healthy volunteers (n=3) into EDTA as anticoagulant (‘E’: BD Vacutainer® EDTA Tube, Catalogue Number: 366643), a related EDTA containing tube with a dense polymer gel base layer which upon centrifugation partitions cellular bodies into the gel (‘G’: BD Vacutainer® Plasma Preparation Tube PPT™, Catalogue Number: 362788), and an EDTA containing tube with a proprietary cocktail of protease inhibitors (PIs) (‘P’: BD P100™ Blood Collection and Preservation System for Plasma Protein Analysis, Catalogue Number: 366456). The plasma samples were collected from individuals who were a mixture of females/males, aged between 25 and 35, who had no apparent evidence of chronic diseases (inflammation, hypertension, diabetes etc.). Each set of samples was donated simultaneously and centrifuged within 30 min of collection at 2500×g at room temperature for 20 minutes. Blood collection was approved by Macquarie University's Human Ethics Committee (Ref: 5201100388). Plasma was recovered, aliquoted and stored at −140°C. These plasma samples were acquired and approved for a range of proteomic investigation. These samples were also utilized in an independent MRM-based study [26] (Figure 1).

Figure 1. Experimental workflow for comparing the proteome and peptidome stability of plasma samples collected from healthy individuals in three different types of blood collection tubes; (i) spray-dried EDTA (E) (ii) EDTA with polymer gel (G) and (iii) EDTA tubes containing a proprietary cocktail of protease inhibitors (P)
MARS 14 Immunodepletion
Plasma samples were depleted to remove the top 14 high abundance proteins (albumin, IgG, transferrin, haptoglobin, alpha 1-anti trypsin, IgA, fibrinogen, alpha2 macroglobulin, alpha1 acid-glycoprotein, IgM, apolipoprotein AI, apolipoprotein AII, complement C3, and transthyretin) using a multiple affinity removal system (MARS-14) immuno-depletion column (Agilent, CA, USA) [44] according to the manufacturer's instructions. Briefly, triplicate plasma samples (tubes E, G and P) collected from three individuals (sample 1, 2, and 3) were thawed and 130 μL was injected onto a MARS-14 affinity column. The bound fraction (BF) (4 ml) and flow through (FT) fractions (2.5 ml) were enriched with high and lower abundance plasma proteins respectively. The FT fractions were collected and then buffer exchanged into 2D gel buffer using a 5000 Da nominal molecular weight cut off filter (Amicon Ultra-2 mL Centrifugal Filters (MC, Millipore). The final volume of the buffer exchanged samples was 200 ul, with the concentration between 2.08-2.56 ug/ul. The protein concentrations in BF and FT were calculated using Bradford Assay (Sigma) according to the manufacturer’s instructions.
1D SDS PAGE analysis
Efficiency of high abundance protein depletion was analysed by one dimensional SDS-PAGE gel electrophoresis ( NuPAGE™ Novex™ 4-12% Bis-Tris Protein Gels, 1.0 mm, 10-well, Standard: Precision Plus Protein™ Kaleidoscope™ Prestained Protein Standards, value pack 1610395). For each BF and FT, 2.5 and 5 µg of total protein was separated on two 4-15% Criterion™ TGX™ Precast Gels ((4-15%) bis-tris pre-cast gradient gels) with MOPS running buffer (Invitrogen) at 200 volt for 50 min. In both of the gels, Precision Plus Protein™ Unstained Standards were used as molecular weight markers. The gels were then fixed, stained overnight with Sypro Ruby (Invitrogen) and imaged on a Typhoon Trio Variable Mode Laser Imager (GE Healthcare).
2D DIGE gel analysis:
Each of the low abundance protein enriched FT fractions (1E,1G, 1P, 2E, 2G, 2P, 3E, 3G and 3P) were analysed and compared using 2D DIGE gel electrophoresis.
Labelling of proteins with CyDyes (minimal, 3 coloured CyDyes) for 2D DIGE gel:
Labelling was performed according to the instructions provided with the kit (GE Healthcare). Briefly, 50 µg of each protein sample was labelled with 400 pmoles of selected CyDyes by incubating on ice for 30 min in the dark. CyDyes labelling was then quenched by the addition of lysine followed by a further incubation on ice for 10 min. A total of 6 DIGE gels were used in this study. Each DIGE gel contains three CyDyes (i.e., Cy2, Cy3 and Cy5) which allows relative comparison of two samples. Samples were labelled with Cy3 and Cy5 and a mixture of the samples was labelled with Cy2 and used as an internal standard. Gel 1 compared 1E & 1G, gel 2 compared 1G & 1P, gel 3 compared 2E & 2G, gel 4 compared 2G & 2P, gel 5 compared 3E & 3G and gel 6 compared 3G & 3P.
1st dimension- Iso-electric focusing (IEF)
The CyDye labelled protein samples were focused on 18 cm 4-7 linear IPG strips using an Ettan IPGphor 2 (GE Healthcare) and the following profile: 300 volts for 4 hours, linear increase from 300 volts to 8000 volts over 8 hours and finally held at 8000 volts until approximately 120 kVh had been accumulated. Throughout the focusing the current limit was set at 50 μA/strip and the temperature at 20 °C.
2nd dimensional- SDS PAGE and imaging
The focused IPG strips were equilibrated for approximately 2 x 15 min each in equilibration buffer (6 M urea, 3% SDS, 20% glycerol, 1x Tris-HCl buffer) then run in the second dimension using an 8-18% gradient gel. The gels were run at 7 mA/gel overnight at 4℃ followed by 35 mA/gel until the bromophenol blue dye front had just run off the bottom of the gel. Immediately after the run, the 2-D CyDye labelled DIGE gels were scanned using a Typhoon Trio variable mode laser imager at a 100 um resolution with the photomultiplier tube set to 550 V.
Image analysis and statistics
The Images were analysed using Progenesis PG240 software (v2006, Nonlinear Dynamics) for quantitative analysis based on protein spot volumes. Spots were detected using auto spot detection method followed by manual edition where necessary. Progenesis ‘ratiometric’ method for DIGE experiments was applied for normalisation of the data, in which the background was subtracted from the images using the Progenesis background subtraction method prior to quantitative analysis [45-47]. Images were compared based on the average spot volume (n = 3) in each group to determine spots which were differentially expressed using Progenesis version 2006 software. Relative abundance changed proteins by >1.5-fold were indicated, when protein spots were expressed 1.5-fold differentially at least in two of the three gels, they were considered as differentially expressed protein spots. Blue spots were down-regulated protein spots and red spots were up-regulated protein spots (Figure 2).
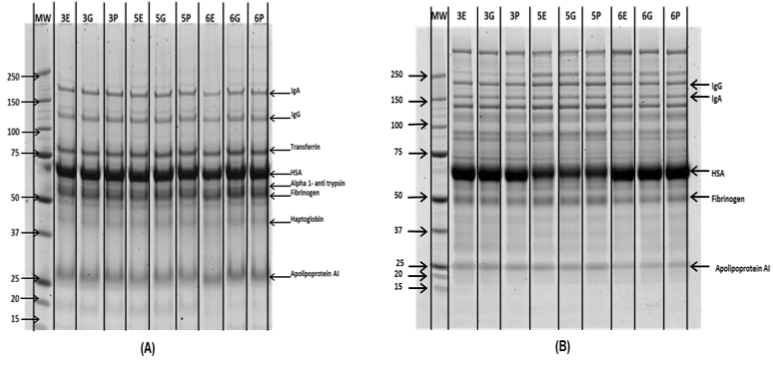
Figure 2. 1Db SDS PAGE analysis of (A) High (BF) and (B) Low abundance (FT) fractions from MARS-14 depletion. Precision Plus Protein™ Unstained Standards were used as molecular marker (MM).
Solid phase extraction
For peptidomic analysis, aliquots of samples 1, 2 and 3 were used. The plasma samples were pooled based on the type of anticoagulant blood collection tube used (E, G and P). This was necessary to obtain the larger volume (>1 ml) that was required to inject enough sample onto the Solid Phase Extraction cartridge. The samples were then passed through a solid phase extraction cartridge (Oasis HLB 1 cc Vac Cartridge (Catalogue No: 186005125, Waters Corp., MA, USA)) according to the manufacturer’s instructions. In detail, the cartridge was conditioned with 1 ml of methanol followed by 1 ml of water. 1 ml of plasma sample was then loaded onto the SPE cartridge. The cartridge was subsequently washed with 1 ml of 5% methanol in water. Finally, the peptides were eluted with 2% formic acid in methanol and 5% NH4OH in methanol (1 ml). The eluates were then dried in a SpeedVac concentrator and resuspended in 1% Trifuloroacetic acid.
MS analysis
An Eksigent UPLC system (AB SCIEX) consisting of a nanoLC 425 UPLC pump and a nanoLC 400 Autosampler was used for delivery of mobile phases and injection of samples. MS data was collected on an Orbitrap Elite (Thermo Fisher Scientific) Mass Spectrometer equipped with a PicoView 550 Nanospray Source (New Objectives). The trap column and analytical column were 100 µm x 3.5 cm and 100 µm x 20 cm respectively which both were packed in-house with Halo® 2.7 µm 160 Å ES-C18 (Advanced Materials Technology). Loading solvent was 0.1% formic acid/(FA) 2% acetonitrile (ACN), analytical solvents A and B were 0.1% FA/5% dimethyl sulfoxide (DMSO) and 0.1% FA/5% DMSO/94.9% ACN.
Following concentration and purification on a C18 Zip Tip (Merck-Millipore), samples were resuspended in 0.1% FA (10 µl) and loaded onto the trap column at a flow of 2.5 µl/min for 10 min. Peptides were eluted onto the analytical column with a linear gradient of solvent B (2.5–12.2%) for 5 min followed by (12.2-80.1%) for 75 min. The flow rate was 450 nL/min and the column temperature 60°C. Each of the prepared samples was analysed three times (n=3) and the LC-MS data for technical replicate (n=3) were acquired individually.
The LC eluent was ionized by applying an electrospray potential of 3.5 KV via a liquid junction connection. The peptide precursors were scanned from 300 to 2400 m/z at 240 k resolution at an AGC target of 1E6 and maximum injection time of 200 ms. Tandem MS performed on the 8 most intense detected ions at the preceding survey scan with CID and HCD fragmentations. The dynamic exclusion duration was set to 40 seconds with 2 repeat counts and 20 seconds duration. Early expiration was enabled. Only precursors with charge state equal to 2 or more were subjected to MS/MS analysis. CID fragmentation spectra were acquired by using the following settings: mass range: normal, scan rate: normal, minimum threshold signal (counts): 500, isolation width: 2Th, normalized collision energy: 35%, activation q: 0.250, activation time: 10 ms, used mass range: 350-1400. HCD fragmentation spectra were acquired by using the following settings: mass range: normal, resolution: 15K, minimum threshold signal (counts): 500, normalized collision energy: 35%, isolation width: 2Th, activation time: 0.1 ms, used mass range: 350-1400.
Peptide identification
The raw data files from Thermo Xcalibur (Thermo Scientific) were imported into Proteome Discoverer version 1.3 (Thermo Fisher Scientific, West Palm Beach, FL, USA,). The data was processed using Mascot v.2.4.0 (Matrix Science, London, UK) with the following criteria: Protein database: Swissprot with decoy database. Enzyme: None. Taxonomy: Homo sapiens. Maximum missed cleavage sites: 9. Precursor mass tolerance: 10 ppm. Fragment mass tolerance: 0.5 Da. Fixed modifications: Oxidation (M), Acetyl (Protein- N term), Amidated (Protein C-term), Phospho (ST), Phospho (Y), Oxidation (HW). Precursor selection: Use MS1 Precursor. Precursor mass range: 700-7000 Da. Total intensity threshold: 50. Minimum peak count: 5. Target FDR (strict): 0.01. Target FDR (Relaxed): 0.05, Query cover= 100%, Identity= 100%.
The list of high confidence peptides from each search were manually analysed and the common peptides from technical replicates are summarised in Table 1. Common peptides that appear in EDTA, Gel EDTA and P100 tubes are compiled in Table 2.
Table 1. Common endogenous peptides identified in plasma samples (biological triplicates) collected in blood collection tubes E, G and P.
Peptide Sequence |
Description |
UniProt ID |
ADSGEGDFLAEGGGVR |
Fibrinogen alpha chain |
P02671 |
GLEEELQFSLGSK |
Complement C4-A |
P0C0L4 |
DSGEGDFLAEGGGVR |
Fibrinogen alpha chain |
P02671 |
NGFKSHALQLNNR |
Complement C4-A |
P0C0L4 |
ADSGEGDFLAEGGGVRGP |
Fibrinogen alpha chain |
P02671 |
NQEQVSPLTLLKLGN |
Alpha-2-antiplasmin |
P08697 |
PGSSGTGGTATWKPGSSGP |
Fibrinogen alpha chain |
P02671 |
SEETKENEGFTVTAEGK |
Complement C3 |
P01024 |
MEPLGRQLTSGP |
Alpha-2-antiplasmin |
P08697 |
KVEQAVETEPEPELR |
Apolipoprotein E |
P02649 |
GSTGNRNPGSSGTGGTATWKPGSSGP |
Fibrinogen alpha chain |
P02671 |
MARS 14 immuno depletion
For immunodepletion through Mars 14 affinity column, 130 ul (conc. 59-61 mg/ ml) of plasma samples were injected (Total protein amount 7700- 7920 ug) while 100 ul of flow through fractions (enriched with low abundance proteins) were collected (conc. 3.51-4.28 mg/ ml, Total protein amount 726-856 ug) which indicates depletion of almost 89-90% of injected proteins (Please refer to Supplementary Table 1 for a complete table of protein concentrations before and after depletion). The depletion chromatograms (Supplementary Figure 1) for plasma samples (1E, 1G, 1P, 2E, 2G, 2P, 3E, 3G and 3P) also show efficient separation of high and low abundance proteins in bound and flow through fractions respectively.
1D SDS PAGE analysis
The bound and flow through fractions (enriched with high and low abundance proteins respectively) from the Agilent MARS-14 immuno-affinity column were analysed by 1D gel electrophoresis. Figure 2 shows the 1D gel image analysis which indicates no major difference in high or low abundance protein abundance between each sample in each biological replicate. The enrichment of low abundance proteins was visible through the separation pattern on 1D gel bands.
2D DIGE analysis
The effect of anticoagulant was evaluated by analysing the low abundance fractions from biological triplicates using 2D DIGE analysis. Visual inspection showed that the general pattern of gel spots between the three anticoagulant treatments in each replicate was similar. In sample 1, a total of 1152 spots were identified. Only 2 spots were differentially abundant between sample 1G and 1P, while there was only one spot identified to be differentially abundant between sample 1E and 1P (Figure 3A). 2D DIGE analysis of sample 2 detected 1513 spots amongst which 11 spots were identified as being differentially abundant between sample 2E and 2G. In contrast, 6 spots were identified to be different between sample 2E and 2P (Figure 3B). Finally, the 2D DIGE analysis of sample 3 identified 1592 different protein spots, where 3 spots were differentially abundant between sample 3E and 3G, 4 spots were different between sample 3G and 3P, and 10 spots were different between sample 3E and 3P (Figure 3C).

Figure 3. 2D DIGE analysis of MARS-14 immunodepleted plasma samples (enriched with low abundance proteins) in biological triplicate (sample 1, 2 and 3) treated with three different varieties of EDTA plasma collection tube (E, G and P) on 6 different gels (Sample 1, 2 and 3 have been analysed on gel 1 & 2, gel 3 & 4 and gel 5 & 6 respectively). Spots circled with red or blue represent proteins that are differentially expressed between two samples on the same gel (Red and blue indicate up and down regulation) and the texts represent the differential expression values.
Peptidomic analysis
The MS results (MS Chromatograms in Supplementary Figure 2 from the peptidomic analysis identified a total of 212 endogenous peptides in all plasma samples that matched to 52 proteins (Supplementary Table 2). Complete independent analysis of EDTA, gel EDTA and P100 samples identified 99, 134 and 60 endogenous peptide sequences that matched with 34, 34 and 20 proteins respectively in each group.
Further analysis of EDTA replicates showed that only 55 peptides were identified in all three replicates, matching to 14 specific proteins (Supplementary Table 3). Similarly, 35 peptides were detected in gel EDTA triplicates that matched to 15 proteins (Supplementary Table 4), where 11 proteins were common (Complement C3, Insulin-like growth factor II, Apo lipoprotein E, Fibrinogen alpha and beta chain, Alpha-2-HS-glycoprotein, Apolipoprotein A-IV, Alpha-2-antiplasmin, Complement C4-A, Inter-alpha-trypsin inhibitor heavy chain H4 and GRB2-associated and regulator of MAPK protein-like OS). In P100 triplicates, 18 peptides were identified that matched with only 8 proteins (Supplementary Table 5). When all the results were compared, only 11 peptides were identified that were common to every plasma sample analysed, matching to 6 proteins (Fibrinogen alpha chain, Complement C4-A, Complement C4-B, Alpha-2-antiplasmin, Complement C3 and Apolipoprotein E).
A number of studies have been conducted to evaluate the use of anticoagulants for preservation of plasma in a suitable state for down-stream analysis. Randall et al. employed a selected reaction monitoring (SRM) strategy and monitored the quantitative variability of plasma proteins collected from seven different individuals in EDTA, gel EDTA and P100 tubes [26]. The study suggested that the protocol for blood processing resulted in only minor differences in the levels of intact plasma proteins that were selected based on their previously known associations with fibrinolysis, coagulation, inflammation or cancer. This study investigated only high abundance proteins like Transthyretin, Kininogen I and Apo lipoprotein A-I. In a separate study, Aguilar-Mahecha et al. investigated the effect of different processing protocols as well as delays in tube processing on the levels of 55 mid and high abundance plasma proteins using MRM-MS assays as well as 27 low abundance cytokines using a commercially available multiplexed bead-based immunoassay. They showed that P100 tubes containing protease inhibitors only conferred proteolytic protection for 4 cytokines. Although mid and high abundance proteins measured by MRM are highly stable in plasma and were left unprocessed for up to six hours, despite the fact that platelet activation can also impact on the levels of these proteins [48]. O’Neil et al. conducted a pilot study using a subset of 24 subjects and compared serum, plasma, and EDTA plasma with proteinase inhibitors for 105 analytes. Although there were notable differences in detectability and measurability between serum and plasma, the EDTA plasma and P100 plasma exhibited similar performance for the majority of analytes, with only eight analytes showing modestly increased CV in P100 plasma [27].
In this study, we aimed to complement the results from Randall et al. by analysing the differential expression of low abundance proteins and endogenous peptides. Therefore, our samples were initially immunodepleted using MARS-14 affinity based system [49,50] to remove the top 14 high abundance proteins (albumin, IgG, transferrin, haptoglobin, alpha 1-anti trypsin, IgA, fibrinogen, alpha2 macroglobulin, alpha1 acid-glycoprotein, IgM, apolipoprotein AI, apolipoprotein AII, complement C3, and transthyretin). The depletion chromatograms indicate efficient and consistent removal of high abundance proteins in the BF, which is further confirmed by separate 1D gel electrophoresis analysis of the BF and FT fractions.
The FT fractions indicated noticeable increase in protein bands that confirmed an effective depletion of high abundance proteins in the FT fraction. The immunodepletion was performed using a MARS-14 immunodepletion column that is commercially available for depletion of high abundance proteins. This column is widely adopted by the research community to reduce the concentration of high abundance proteins and enrich the low abundance protein fractions in highly complex plasma samples. A number of studies have strongly suggested that commercial MARS-14 antibody affinity columns that claims to selectively deplete 14 high abundance proteins present in plasma offer efficient, specific and reproducible depletion [49,50]. Therefore, we decided to primarily focus on the depleted plasma fractions that were enriched with low abundance proteins. In the subsequent experiment, 2D DIGE was applied to investigate the differential expression of low abundance proteins between three different anticoagulant pre-treatments. EDTA, gel EDTA and P100 pre-treated plasma showed very few changes in the recovery and expression pattern between three biological replicates (3 out of 1152, 17 out of 1513 and 17 out of 1592 spots in sample 1, 2 and 3 respectively) and were not consistent across three different anticoagulant treatments. These findings support the hypothesis of Randal et al. that there is no evidence of reproducible change in low abundance protein expression I irrespective of the anticoagulant used in the blood collection tube [26].
When conducting a peptidomic study, it is important that the endogenous peptides remain stable in plasma for downstream biomarker analysis. The role of peptidase activity in several phases of the peptide lifecycle (including the production, activation, inactivation and degradation of bioactive peptides) cannot be ignored [51]. While some of these peptide-peptidase pairings are well understood, there are still a vast number of bioactive peptides whose in vivo regulation by peptidases have not been completely characterized [51]. Yi et al. have examined time dependent changes in enriched endogenous plasma peptides using MALDI-MS and claimed that inclusion of protease inhibitors into the blood collection tube can alleviate peptidase-related preanalytical variability [52]. This finding supports the previous claims where protease inhibition has been suggested to stabilise the presence of endogenous plasma peptides when samples have been maintained at sub-optimal temperatures for extended time periods [53].
Our data from peptidomic analysis indicates the number of endogenous peptides (n=18) present in P100 treated triplicate samples is almost 33% less than the number of peptides found in EDTA treated triplicates (n= 55) and 50% of the number of peptides found in Gel EDTA treated triplicates (n=35). Interestingly, 11 peptides were identified in all the replicates that match to 6 protein identifications (namely Fibrinogen alpha chain, Complement C4-A, Complement C4-B, Alpha-2-antiplasmin, Complement C3 and Apolipoprotein E). Therefore it can be suggested that the above six proteins can degrade upon proteolysis in the plasma samples post collection, during storage and release endogenous peptides, irrespective of the anticoagulant used. It was interesting to note that, although endogenous peptides derived from serum albumin (the most abundant plasma protein that comprises approx. 70% of total plasma proteins) were identified, there were other endogenous peptides that were derived from three other high abundance plasma proteins (Fibrinogen alpha chain, Complement C3 and Apolipoprotein E). However it is also evident that endogenous peptides are released from medium and low abundance proteins like complement C4 and Alpha-2-antiplasmin in all the EDTA tube varieties.
Careful analysis showed, only EDTA treated samples contained endogenous peptides derived from two proteins (Alpha-2-macroglobulin and Serum albumin) that were not found in Gel EDTA or P100 treated samples. In contrast, gel EDTA and P100 treated samples had endogenous peptides from four (Apolipoprotein C-III, Apolipoprotein C-II, Transthyretin and Extracellular matrix protein 1) and one (Alpha-1-antichymotrypsin) protein respectively that were not found in EDTA samples. Moreover, it was also noted that EDTA treated plasma samples contained endogenous peptides derived from 8 medium and low abundance proteins, (namely Alpha-2-antiplasmin, Alpha-2-HS-glycoprotein, Apolipoprotein E, Complement C4-A, Complement C4-B, GRB2-associated and regulator of MAPK protein-like, Insulin-like growth factor II, Inter-alpha-trypsin inhibitor heavy chain H4, and Transthyretin), 5 of which are common to the endogenous peptides found in gel EDTA. The gel EDTA treated samples had endogenous peptides derived from 7 low abundance proteins (namely Alpha-2-antiplasmin, Alpha-2-HS-glycoprotein, Extracellular matrix protein 1, Insulin-like growth factor II, Inter-alpha-trypsin inhibitor heavy chain H4 and Transthyretin). In contrast, samples collected in P100 tube had endogenous peptides collected from only three medium and low abundance proteins (Alpha-1-antichymotrypsin, Alpha-2-antiplasmin and complement C4). Our results further show that endogenous peptides derived from Alpha-1-antichymotrypsin were found only in protease inhibitor containing tubes and there is no reproducible evidence of endogenous peptides derived from Alpha-1-antichymotrypsin in samples treated with EDTA and gel EDTA.
As the results were further analysed, it was expected to find Alpha-1-antichymotrypsin in plasma treated with protease inhibitors, as this protein inhibits the activity of proteases in human body [54]. As an acute phase protein, Alpha-1-antichymotrypsin is associated with prostate cancer, liver disease, Parkinson’s disease, Alzheimer’s disease and chronic obstructive pulmonary disease [55,56].
Our data shows that
An appropriate standard anticoagulant in the blood collection tubes is essential for the reproducible and reliable discovery and validation of plasma biomarker using proteomic studies. EDTA is the cheapest and most commonly used anticoagulant used in blood collection tubes for clinical and diagnostic purposes. Our observations strongly suggest that plasma samples collected in blood collection tubes containing EDTA are suitable for conducting proteomics investigation on clinical samples compared to more expensive blood collection tubes containing protease inhibitors that are specifically designed for research purposes. This study also indicates that the tubes containing protease inhibitors can occasionally be considered as a suitable option for preserving samples for downstream peptidomic analysis. As the peptidomic study has been done using a pool of plasma samples based on the anticoagulant used, we cannot ignore the fact that individual biological variation can create observational errors. Nevertheless the type of blood collection tubes and anticoagulants used will be an essential requirement of all SOPs generated for routine sample collection and handling.
This study was supported with project funding from the Australian National Health and Medical Research Council (NHMRC APP1010303), NSW Cancer Council (RG10-04) and the Cancer Institute NSW (15/ECF/1-38) and supported through the Department of Biomedical Sciences, Macquarie University. Some of the research described herein was facilitated by access to the Australian Proteome Analysis Facility (APAF) established under the Australian Government’s National Collaborative Research Infrastructure Strategy (NCRIS).
Supplementary Table 271016
- Hsieh SY, Chen RK, Pan YH, HL (2006) Systematical evaluation of the effects of sample collection procedures on low-molecular-weight serum/plasma proteome profiling. Proteomics 2006; 6: p. 3189-98. [Crossref]
- Shen Y, Liu T, ToliÄ N, Petritis BO, Zhao R, et al. (2010) Strategy for degradomic-peptidomic analysis of human blood plasma. J Proteome Res 9: 2339-2346. [Crossref]
- Hanash S (2012) A call for a fresh new looks at the plasma proteome. Proteomics Clin Appl 6: 443-446. [Crossref]
- Anderson NL, NG Anderson (2002) The human plasma proteome history, character and diagnostic prospects. Molecular & Cellular Proteomics 2002; 1(11): p 845-867. [Crossref]
- Lundblad R (2005) Considerations for the use of blood plasma and serum for proteomic analysis. The Internet Journal of Gastroenterology 2005; 1(2).
- Karczmarski J, Rubel T, Mikula M, Wolski J, Rutkowski A (2013) Pre-analytical-related variability influencing serum peptide profiles demonstrated in a mass spectrometry-based search for colorectal and prostate cancer biomarkers. Acta Biochimica Polonica 2013; 3(60): p 417-425. [Crossref]
- Kushnir MM (2013) Are samples in your freezer still good for biomarker discovery? American journal of clinical pathology 2013; 140(3): p. 287-288. [Crossref]
- Percy AJ, Parker CE, Borchers CH (2013) Pre-analytical and analytical variability in absolute quantitative MRM-based plasma proteomic studies. Bioanalysis 2013; 5(22): p. 2837-56. [Crossref]
- Fung KY, Nice E, Priebe I, Belobrajdic D, Phatak A (2014) Colorectal cancer biomarkers: To be or not to be? Cautionary tales from a road well-travelled. World journal of gastroenterology: WJG 2014; 20(4): p. 888. [Crossref]
- Zhang H, Liu Q, Zimmerman LJ, Ham AJ, Slebos RJ, et al. (2011) Methods for peptide and protein quantitation by liquid chromatography-multiple reaction monitoring mass spectrometry. Mol Cell Proteomics 10: M110. [Crossref]
- Surinova S, Schiess R, Hüttenhain R, Cerciello F, Wollscheid B, et al. (2011) On the development of plasma protein biomarkers. J Proteome Res 10: 5-16. [Crossref]
- Luchansky MS, Washburn AL, McClellan MS, Bailey RC (2011) Sensitive on-chip detection of a protein biomarker in human serum and plasma over an extended dynamic range using silicon photonic microring resonators and sub-micron beads. Lab Chip 2011; 11(12): p. 2042-4. [Crossref]
- Merrick BA (2003) The human proteome organization (HUPO) and environmental health. EHP Toxicogenomics 111: 1-5. [Crossref]
- Omenn GS (2004) Advancement of biomarker discovery and validation through the HUPO plasma proteome project. Dis Markers 20: 131-134. [Crossref]
- Omenn GS (2004) International collaboration in clinical chemistry and laboratory medicine: the Human Proteome Organization (HUPO) Plasma Proteome Project. Clin Chem Lab Med 2004; 42(1): p. 1-2.
- Muthusamy B, Hanumanthu G, Suresh S, Rekha B, Srinivas D, et al. (2005) Plasma Proteome Database as a resource for proteomics research. Proteomics 5: 3531-3536. [Crossref]
- Omenn GS (2004) Advancement of biomarker discovery and validation through the HUPO plasma proteome project. Dis Markers 20: 131-134. [Crossref]
- Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Schuchard MD, et al. (2005) HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 2005; 5(13): p. 3262-3277. [Crossref]
- Hildebrand D, Merkel P, Eggers LF, Schlüter H (2013) Proteolytic processing of angiotensin-I in human blood plasma. PLoS One 8: e64027. [Crossref]
- Wildes D, Wells JA (2010) Sampling the N-terminal proteome of human blood. Proc Natl Acad Sci U S A 2010; 107(10): p. 4561-6. [Crossref]
- Xiao-YD, Brian AZ, Timothy M, Samantha JA, Tracy MH, et al. (2009) Regulation of chemerin bioactivity by plasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activable fibrinolysis inhibitor), and platelets. J Biol Chem 2009; 284(2): p751-8. [Crossref]
- Luque-Garcia JL, Neubert TA (2007) Sample preparation for serum/plasma profiling and biomarker identification by mass spectrometry. J Chromatogr A 2007; 1153(1-2): p 259-76. [Crossref]
- Drake SK, Bowen RA, Remaley AT, Hortin GL (2004) Potential interferences from blood collection tubes in mass spectrometric analyses of serum polypeptides. Clin Chem 50: 2398-2401. [Crossref]
- Shen Y, ToliÄ N, Liu T, Zhao R, Petritis BO, et al. (2010) Blood peptidome-degradome profile of breast cancer. PLoS One 5: e13133. [Crossref]
- Randall SA, McKay MJ, Molloy MP (2010) Evaluation of blood collection tubes using selected reaction monitoring MS: implications for proteomic biomarker studies. Proteomics 10: 2050-2056. [Crossref]
- O'Neal WK, Anderson W, Basta PV, Carretta EE, Doerschuk CM, et al. (2014) Comparison of serum, EDTA plasma and P100 plasma for luminex-based biomarker multiplex assays in patients with chronic obstructive pulmonary disease in the SPIROMICS study. J Transl Med 12: 9. [Crossref]
- Yi J, Craft D, Gelfand CA (2011) Minimizing preanalytical variation of plasma samples by proper blood collection and handling. Methods Mol Biol728: p. 137-49. [Crossref]
- Rai AJ, Vitzthum F (2006) Effects of preanalytical variables on peptide and protein measurements in human serum and plasma: implications for clinical proteomics. Expert review of proteomics 2006; 3(4): p. 409-426. [Crossref]
- Barelli S, Crettaz D, Thadikkaran L, Rubin O, Tissot JD (2007) Plasma/serum proteomics: pre-analytical issues. Expert Rev Proteomics 2007; 4: 363-370. [Crossref]
- Banks RE, Stanley AJ, Cairns DA, Barrett JH, Clarke P, et al. (2005) Influences of blood sample processing on low–molecular-weight proteome identified by surface-enhanced laser desorption/ionization mass spectrometry. Clinical chemistry, 2005; 51(9): p. 1637-1649. [Crossref]
- Nanjappa V, Thomas JK, Marimuthu A, Muthusamy B, Radhakrishnan A, et al. (2014) Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res 42: D959-965. [Crossref]
- Kim HJ, Kim MR, So EJ and Kim CW (2007) Comparison of proteomes in various human plasma preparations by two-dimensional gel electrophoresis. J Biochem Biophys Methods 70: 619-625. [Crossref]
- Rossignol P, Cambillau M, Bissery A, Mouradian D, Benetos A, et al. (2008) Influence of blood sampling procedure on plasma concentrations of matrix metalloproteinases and their tissue inhibitors. Clin Exp Pharmacol Physiol 2008; 35(4): p. 464-9. [Crossref]
- Krakow EF, Goudar R, Petzold E, Suvarna S, Last M, et al. (2007) Influence of sample collection and storage on the detection of platelet factor 4-heparin antibodies. Am J Clin Pathol 128: 150-155. [Crossref]
- Mohri M, Rezapoor H (2009) Effects of heparin, citrate, and EDTA on plasma biochemistry of sheep: comparison with serum. Res Vet Sci 86: 111-114. [Crossref]
- Banfi G, Salvagno GL, Lippi G (2007) The role of ethylenediamine tetraacetic acid (EDTA) as in vitro anticoagulant for diagnostic purposes. Clin Chem Lab Med 45: 565-576. [Crossref]
- Yi J, C Kim, Gelfand CA (2007) Inhibition of intrinsic proteolytic activities moderates preanalytical variability and instability of human plasma. Journal of proteome research 2007; 6(5): p. 1768-1781. [Crossref]
- Adriana AM, Michael AK, Dominik D, Christoph HB, Mark B (2012) The effect of pre-analytical variability on the measurement of MRM-MS-based mid-to high-abundance plasma protein biomarkers and a panel of cytokines. PLoS One 2012; 7(6): p. e38290-e38290. [Crossref]
- Marouga RS, David, Hawkins E (2005) The development of the DIGE system: 2D fluorescence difference gel analysis technology. Analytical and bioanalytical chemistry 2005; 382(3): p. 669-678. [Crossref]
- Palsuledesai CC, Ochocki JD, Markowski TW, Distefano MD (2014) A combination of metabolic labeling and 2D-DIGE analysis in response to a farnesyltransferase inhibitor facilitates the discovery of new prenylated proteins. Mol Biosyst 2014; 10(5). [Crossref]
- Mahboob S, Mohamedali A, Ahn SB, Schulz-Knappe P, Nice E, et al. (2015) Is isolation of comprehensive human plasma peptidomes an achievable quest? J Proteomics 127: 300-309. [Crossref]
- Tu C, Rudnick PA, Martinez MY, Cheek KL, Stein SE, et al. (2010) Depletion of abundant plasma proteins and limitations of plasma proteomics. J Proteome Res 9: 4982-4991. [Crossref]
- Ahn SB, Khan A (2014) Detection and quantitation of twenty-seven cytokines, chemokines and growth factors pre-and post-high abundance protein depletion in human plasma. EuPA Open Proteomics 2014; 3: p. 78-84.
- Fernando N, Panozzo J, Tausz M, Norton R, Fitzgerald G, et al. (2015) Rising CO 2 concentration altered wheat grain proteome and flour rheological characteristics. Food chemistry 170: 448-454. [Crossref]
- Polaskova V, Kapur A, Khan A, Molloy MP, Baker MS (2010) High-abundance protein depletion: Comparison of methods for human plasma biomarker discovery. Electrophoresis 31: 471-482. [Crossref]
- Aguilar MA, Kuzyk MA, Domanski D, Borchers CH, Basik M (2012) The effect of pre-analytical variability on the measurement of MRM-MS-based mid- to high-abundance plasma protein biomarkers and a panel of cytokines. PLoS One 7: e38290. [Crossref]
- Cao TH, Quinn PA, Sandhu JK, Voors AA, Lang CC, et al. (2015) Identification of novel biomarkers in plasma for prediction of treatment response in patients with heart failure. Lancet 385 Suppl 1: S26. [Crossref]
- Liu Y, Buil A, Collins BC, Gillet LC, Blum LC, et al. (2015) Quantitative variability of 342 plasma proteins in a human twin population. Mol Syst Biol 11: 786. [Crossref]
- Lone AM, Kim YG, Saghatelian A (2013) Peptidomics methods for the identification of peptidase–substrate interactions. Current Opinion in Chemical Biology 17: 83-89. [Crossref]
- Yi J, Liu Z, Craft D, O'Mullan P, Ju G, et al. (2008) Intrinsic peptidase activity causes a sequential multi-step reaction (SMSR) in digestion of human plasma peptides. J Proteome Res 2008; 7(12): p. 5112-8. [Crossref]
- Waters T, Coated E (2007) Inhibition of Intrinsic Proteolytic Activity Moderates Preanalytical Variability and Stabilizes the Human Plasma. Proteome. J Proteome Res. [Crossref]
- Matsubara E (1990) A1-Antichymotrypsin as a possible biochemical marker for Alzheimer-type dementia. Annals of neurology 28: 561-567.
- Christensson A, Björk T, Nilsson O, Dahlén U, Matikainen MT, et al. (1993) Serum prostate specific antigen complexed to alpha 1-antichymotrypsin as an indicator of prostate cancer. J Urol 150: 100-105. [Crossref]
- Stenman UH, Leinonen J, Alfthan H, Rannikko S, Tuhkanen K, et al., (1991) A complex between prostate-specific antigen and a1-antichymotrypsin is the major form of prostate-specific antigen in serum of patients with prostatic cancer: assay of the complex improves clinical sensitivity for cancer. Cancer Research 51: 222-226. [Crossref]