This article reviews the main body of knowledge regarding NHE1 and NHE3 exchangers and their interaction with Angiotensin II, Angiotensin-(1-7), Aldosterone and Arginine Vasopressin, particularly their renal actions. This work addresses the biphasic effects of different hormonal doses on NHE1 or NHE3 in proximal tubule in Wistar, SHR (hypertensives) and their control WKY (normotensives) rats or MDCK cells (which share similarities with the collecting duct). The hormones were applied alone, with their inhibitors or plus agents that change the [Ca2+]i. The data are compatible with hormonal stimulation of these exchangers by increases of [Ca2+]i in lower range, and inhibition at high [Ca2+]i. In MDCK cells and Wistar rats, low doses of ANG II, ALDO or AVP stimulated the exchangers, while high doses inhibited them. ANG–(1-7), in Wistar or WKY rats has inverse, dose-dependent effects. In SHR rats, the biphasic effects of ANG-(1-7) were similar to the effects of ANG II, ALDO or AVP in Wistar rats. The interactions between these effects may represent a mechanism that regulates extracellular volume. In hypertensives animals, a high plasma level of ANG-(1-7) inhibited NHE3 in the proximal tubule, which mitigated hypertension. Figure 6 shows a schematic model to describe these biphasic hormonal effects.
NHE
The Na+/H+ exchanger (NHE) is a ubiquitous membrane protein that is present as a number of isoforms in living organisms. In mammals, the NHE1 and NHE3 isoforms catalyze the electroneutral exchange of Na+ and H+ to control their respective concentration gradients, a process that is essential for numerous physiological processes, including controlling cell volume, pH, and systemic electrolyte levels, and acid-base and fluid volume homeostasis. In addition, NHE activity facilitates the progress of other cellular events, such as adhesion, migration, and proliferation. So far, ten different mammalian NHE isoforms (NHE1-10) that share 25-70% amino acid identity have been identified and characterized [1-3]. These isoforms have a common predicted secondary structure that consists of 12 conserved membrane-spanning segments at the amino-terminus and a more divergent, cytoplasmically oriented carboxy-terminus. These isoforms show considerable heterogeneity in their patterns of tissue/cell expression and membrane localization. Functional studies have revealed further differences in their kinetic properties, their sensitivity to pharmacological antagonists, their regulation by diverse hormonal and mechanical stimuli, and many of their essential physiological functions. In the nephron, individual NHE isoforms have different functions. These functions are reflected in their differential expression along the segments of the nephron, their localization in renal epithelial cells at the basolateral (e.g., isoform NHE1) or apical surface (e.g., isoform NHE3), and their activation in response to distinct agonists [4-13].
NHE1
The NHE1 isoform is ubiquitously expressed in the plasma membranes of virtually all mammalian cells, where it regulates intracellular pH, salt concentration and cell volume [14-16]. Therefore, NHE1 is critical for controlling and maintaining some of the most fundamental processes in cellular physiology, including cell growth and differentiation [14]. NHE1 has two functional domains: an amino-terminal ion translocation domain that consists of ~500 amino acids that catalyzes amiloride-sensitive Na+/H+ exchange and contains a built-in modifier site (pH sensor) and a regulatory carboxy-terminal cytoplasmic domain that consists of ~300 amino acids that determines the set point value of the modifier site [17,18]. Depending on the stimulus, NHE1 activation is often associated with one of two mechanisms: 1) phosphorylation, such as by assorted serine/threonine protein kinases [19], p38 mitogen-activated protein kinase (MAPK) [20] and p90 ribosomal S6 kinase [21], and 2) binding to regulatory proteins, resulting in conformational changes. Specifically, phosphatidylinositol 4,5-bisphosphate (PIP2) binds to the juxtamembrane region of NHE1, and actin-binding proteins in the ezrin, radixin and moesin family (ERM) connect NHE1 to the cytoskeleton. Different serine kinases, such as the ERK-regulated kinase p90RSK, the Ste20-like Nck-interacting kinase (NIK) and the Rho-associated kinase p160ROCK, phosphorylate NHE1 near its C-terminus [22]. In addition, Ca2+ regulates NHE1 by binding to its juxtamembrane region via calcineurin B homologous proteins 1 and 2 (CHP1 and CHP2) [23,24] and tescalcin (CHP3) [25] or by binding to two neighboring sites in its C-terminal regulatory domain via calmodulin (CaM) [26]. Associations with its high-affinity CaM domain release an autoinhibitory intramolecular interaction that enhances NHE1 activity [11]. Although several other studies have been conducted to clarify how binding between Ca2+/CaM and NHE1 occurs [27-31], and while the structure of the juxtamembrane region of the regulatory domain (503-545) involved in complexes with CHP1 or CHP2 has been determined using nuclear magnetic resonance [32] or X-ray crystallography [33], the binding mechanism by which Ca2+/CaM activates NHE1 still is not well-described. Recently, using small-angle X-ray scattering analysis, Köster and co-workers [1] studied the molecular mechanisms underlying the phosphorylation-dependent regulation of NHE1, and they proposed an extended model to explain how Ca2+ regulates NHE1 activity via its C-terminal regulatory domain. These authors showed how CaM interacts with both CaM-binding regions in NHE1 and provided insight into how posttranslational modification by phosphorylation affects CaM binding to results in either the stimulation or the inhibition of NHE1 activity. In this model, CaM binding weakened the interaction between the autoinhibitory region and the proton modifier site, allowing protons unhindered access to this site and resulting in the up-regulation of the transport activity of NHE1. These authors [1] also reported that: 1) upon CaM binding, NHE1 is activated by a shift in sensitivity towards an alkaline intracellular pH, 2) the 2.23 Å crystal structure of the NHE1 CaM binding region (NHE1CaMBR) complexes with CaM and intracellular calcium [Ca2+]i, 3) the C- and N-lobes of CaM bind to the first and second helix of NHE1CaMBR, respectively, and 4) both the NHE1 helices and Ca2+ -bound CaM become elongated, as confirmed by an analysis of their X-ray structure. More recently, it was demonstrated that 1) NHE1 and CaM are associated in vivo through endothelin-dependent signaling pathways [7], and 2) the modulation of NHE1 activity by various activators and inhibitors occurs as a result of the direct binding of these molecules to the lipid-interacting domain (LID), which alters the association between the LID and the plasma membrane [8].
NHE3
The NHE3 isoform is present in the epithelial brush borders of intestinal Na+-absorptive cells and in renal tubules, where it mediates the majority of gastrointestinal and renal Na+ absorption [34,35] and renal HCO3- reabsorption [36-38]. NHE3 also influences other brush border transport processes, such as intestinal brush border Cl−/HCO3− exchanger, a putative anion transporter [39,40], and Cl− secretion, which is mediated by the cystic fibrosis transmembrane regulator [41,42]. The more consistent characteristics of NHE3 include the following: 1) it is activated under basal conditions, 2) under basal conditions, NHE3 interacts with Ca2+/calmodulin-dependent protein kinase II (CaMKII), 3) only the γ isoform of this kinase associates with NHE3, 4) the NHE3 C-terminal domain, which is necessary for CaMKII binding under basal conditions, is 586–605 aa long and was predicted using multiple modeling programs to be α-helical, and 5) binding is rapidly reduced under conditions involving elevated physiological levels of Ca2+ [13]. However, other proteins associate with NHE3, including NHERF1–4, phospholipase Cγ, and CK2α [43, 44]. In addition, the CaMKII-mediated inhibition of basal NHE3 activity is NHERF2-dependent, occurs as a result of changes in the NHE3 turnover number and is associated with the phosphorylation of NHE3. This regulatory effect requires amino acids at its C-terminal to interact with the CaMKII binding domain downstream of NHE3 aa 690 (Ser693, Ser694, and Ser810), which is part of the putative CaMKII phosphorylation consensus sequence [45,46]. Recently, it was demonstrated that 1) NHE3 basal activity is regulated by a signaling complex that is controlled by the sequential effects of two kinases, Akt and GSK-3, which act on a Ser cluster in the same NHE3 C-terminal domain that binds ezrin, and 2) these kinases regulate the dynamic association between ezrin and NHE3 to affect basal NHE3 activity [9]. CaMKII is therefore generally inactive in the presence of basal levels of Ca2+, and active CaMKII is generally created via the autophosphorylation and release of the kinase sequence from the CaMKII autoinhibitory domain. These events are followed by autophosphorylation at Thr286/287 (depending on the species being studied) or oxidation at Met281 or Met282 [47], which result in a conformational change in CaMKII that allows high affinity interactions with its target proteins and prevents the inactivation of the kinase by re-association between its catalytic domain with the autoinhibitory domain when Ca2+ returns to basal levels. Similarities in the sequence alignment between the CaMKII binding domain of NHE3 and the CaMKII autoinhibitory domain [48] suggest that the catalytic subunit of the activated CaMKII (freed from its autoinhibitory domain) binds to a domain in its substrate, which resembles the kinase autoinhibitory domain but cannot inactivate the catalytic domain, while preventing access to the kinase autoinhibitory domain [49,50]. Therefore, CaMKII constitutively binds to, phosphorylates, and inhibits NHE3 via a NHERF2 protein-dependent process [48]. Because it has multiple functions, NHE3 is regulated by a wide variety of agonists in response to physiological conditions [4,13,51].
The kidney is an important source of several components of the Renin-Angiotensin System (RAS) as well as a target organ for their activities. These mechanisms were recently described in a relevant review [52]. Most of the major known effects of the RAS are related to the activity of ANG II. However, a growing amount of evidence indicates that other peptides that have been more recently described, such as ANG-(1-7) and their respective receptors, increase the functional spectrum of the RAS [see below in ANG-(1-7) section]. Angiotensinogen, an α-2-globulin that is constitutively produced and released into the circulation mainly by the liver, is converted to ANG I by Renin, and the mRNAs of both ANG I and Renin are expressed in juxtaglomerular cells and renal tubular cells. Angiotensin-converting enzyme, which cleaves ANG I to form ANG II, is found principally in the lung but is also found in other tissues, including the kidneys. Complete RASs have also been described in other tissues, including the brain, vasculature and adrenal cortex [53,54].
In addition to its presence as a circulating hormone that has strong vasoconstrictive effects on systemic and glomerular hemodynamics, ANG II also has important paracrine effects on cell proliferation and the tubular transport of ions [55]. Under pathological conditions, ANG II has been shown to promote vascular remodeling, cardiac hypertrophic remodeling, and extracellular matrix deposition [56], and it is also implicated in inflammation, endothelial dysfunction, atherosclerosis, hypertension, and congestive heart failure.
Receptors and intracellular pathways
In this section, we briefly overview the available data regarding the signaling and functions of the various ANG II receptors. These topics have been reviewed in more detail in other publications [57,58]. There are two major ANG II receptors, AT1 and AT2. In addition, AT1 receptors are further subdivided into AT1A and AT1B [57]. While AT1 may be the main receptor that mediates the effects of ANG II on blood pressure, AT2 may also be partially involved in the regulation of blood pressure [59]. ANG II regulates blood pressure via AT1 receptors in both renal and extrarenal tissues and renal and extrarenal AT1A receptors contribute almost equally to maintaining baseline blood pressure [60,61]. ANG II exerts a wide variety of functions via AT1, including (but not limited to) roles in cardiovascular homeostasis, renal functions, ion flux, protein phosphorylation, gene expression, cell growth, the stimulation of ALDO release, and central effects, such as eliciting thirst and AVP secretion [62].
The AT1 receptor is a 40-kDa protein that consists of 359 amino acids and is a member of the seven transmembrane domain G protein-coupled receptor family.These receptors typically couple with Gq complexes, resulting in the activation of downstream intracellular signaling pathways that lead to the activation of phospholipase C (PLC), phospholipase A2 (PLA2), and phospholipase D (PLD) [63]. The activation of PLC triggers an increase in the formation of 1,4,5-inositol triphosphate (IP3) and diacylglycerol (DAG), which promote the release of calcium from intracellular stores and the activation of protein kinase C (PKC), respectively [58]. In addition to inducing G protein- and non-G protein-related signaling pathways, ANG II, via AT1 receptors, performs its functions via MAP kinases (e.g., ERK 1/2, JNK, and p38MAPK), receptor tyrosine kinases (e.g., PDGF, EGFR, and insulin receptor), and nonreceptor tyrosine kinases [e.g., Src, JAK/STAT and focal adhesion kinase (FAK)]. The AT1R-mediated activation of NAD(P)H oxidase leads to the generation of reactive oxygen species, which are widely implicated in vascular inflammation and fibrosis. ANG II also promotes associations between scaffolding proteins, such as paxillin, talin, and p130Cas, which leads to focal adhesions and extracellular matrix formation. These signaling cascades lead to contraction, smooth muscle cell growth, hypertrophy, and cell migration, which are events that contribute to both normal vascular functions and disease progression [58]. The mechanisms involved in the regulation of the AT1 receptor functions and the magnitude of local ANG II effects include the regulation of the following: 1) AT1 receptor transcription, 2) AT1 receptor surface expression via internalization and membrane recycling and 3) AT1 receptor activity by accessory proteins.
The AT2 receptor is a seven-transmembrane domain protein that consists of 363 amino acids that have a molecular mass of 41 kDa and share 34% homology with AT1 [64]. AT2 receptors are highly expressed in a variety of tissues in developing embryos, but AT2 expression declines after birth [65,66]. Until now, the range of AT2-dependent functions and AT2-linked intracellular signaling events are still not completely understood [67]. In addition, under certain conditions, AT2 receptors form heterodimers with AT1 receptors, which results in the attenuation of AT1-mediated effects on ANG II (1). Several studies confirmed that ANG II binding to AT2 results in an increase in the formation of vasodilator agents, such as NO, prostanoids, and bradykinin [68].
The renal proximal tubule reabsorbs approximately 65% of the NaCl that it filters, which contributes to the regulation of plasma volume and blood pressure. Moreover, the proximal tubule reabsorbs approximately 80% of filtered bicarbonate, which plays an important role in the maintenance of systemic acid-base balance [69]. This process is dependent primarily on Na+, involves the luminal Na+/H+ exchanger (NHE3) and the basolateral Na+-HCO3- cotransporter (NBC) and mediates a majority of sodium-coupled proximal renal reabsorption processes [69,70]. Under normal conditions, ANG II participates in several mechanisms that are involved in the renal acidification process. In their study of the in vivo microperfusion of the rat proximal convoluted tubule, Liu and Cogan were the first to show that ANG II increases proximal tubular bicarbonate reabsorption [71]. Another study also using an in vivo stationary microperfusion technique to study the rat proximal tubule [72] indicated that ANG II added to luminal or peritubular perfusion fluid stimulates bicarbonate reabsorption [73]. The stimulatory effects of ANG II on proximal NHE3 activity are mediated by both cAMP-dependent [71,74] and cAMP-independent mechanisms [75-77], although these effects may depend on the specific ANG II receptor subtype [78]. In addition, it is recognized that ANG II stimulates proximal Na+-HCO3- reabsorption by stimulating basolateral Na+-HCO3- cotransporter activity [79-81]. More recently, other studies have indicated that in IRPTC cells (a SV40-transformed cell line that was derived from rat proximal tubules) long-term exposure to ANG II (10-9 M) resulted in the upregulation of H+-ATPase activity at least in part because it increased the cell surface expression of B2 subunit. These studies have also demonstrated that this regulatory pathway is dependent on mechanisms involving the activation of tyrosine kinase, p38 MAPK, and PI3K.
Dose-dependent biphasic effects of ANG II in the proximal tubule
It has been widely reported that in the proximal tubule, the effects of ANG II on NHE3 may be biphasic and dose-dependent: at low concentration, ANG II stimulates NHE3, while high concentrations of ANG II inhibit NHE3 [82,83]. Electrophysiological studies of isolated and perfused S2 segments of rabbit renal proximal tubules have confirmed that picomolar concentrations of ANG II stimulate [79] and micromolar concentrations of ANG II inhibit basolateral Na+-HCO3- cotransport [63]. The effects of ANG II on Na+/K+-ATPase in the proximal tubule are also biphasic [79,84,85]. It is likely that the inhibitory effect of high concentrations of ANG II on proximal transport could have some physiological significance because intrarenal concentrations of ANG II are much higher than the concentrations observed in plasma [86].
Although controversial data have been reported concerning the receptor subtype(s) that are responsible for the biphasic effects of ANG II on proximal tubular transport [87,88], some studies have clearly indicated that both luminal and basolateral AT1A receptors mediate the biphasic effects of Ang II on proximal transport [89-91]. It has been shown that at physiological concentrations, binding between ANG II and the AT1 receptor induces the rapid activation of PLC, which results in the release of IP3 and DAG, which are themselves involved in slightly increasing [Ca2+]i by mobilizing Ca2+ from intracellular storage and by activating PKC, respectively [92-94]. These processes may ultimately result in the activation of ERK [71,83,90]. At high concentrations, ANG II induces the activation of the phospholipase A2 (PLA2)/arachidonic acid/5,6-epoxyeicosatrienoic acid (EET) pathway and/or the NO/cGMP pathway and the mobilization of intracellular Ca2+, also through mechanisms involving the AT1 receptor [83,86,95,96].
Hence, through these different pathways (which lead to lower or higher levels of [Ca2+]i through changes that are mediated by the AT1 receptor), ANG II exerts a dose-dependent bimodal effect. These findings were confirmed in vitro when it was demonstrated that in rat intact proximal tubule cells, ANG II, at low doses, stimulated NHE3 through a G protein–dependent protein kinase C pathway, whereas at higher doses, ANG II inhibited NHE3 activity through a mechanism involving cytochrome P-450–dependent metabolites [83]. In addition, in rabbit proximal tubular cells, physiological concentrations of ANG II stimulated amiloride-sensitive NHE3 activity [97]. In renal cortical homogenates [98] and cultured renal proximal cells [99], ANG II inhibited adenyl cyclase. More recently, in opossum kidney proximal tubule (OKP) cells, it was confirmed that the activation of NHE3 by ANG II is mediated by IP3 receptor-binding protein, which is released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II [100]. These results support the physiological relevance of IRBIT as an NHE3-interacting protein and its critical role in the regulation of NHE3 activity by ANG II, at least in vitro.
While the biphasic effects of ANG II on proximal tubular transport have been studied in rats, mice, and rabbits [101-105], little is known about the effects of ANG II on human proximal transport. However, recently, Shirai and colleagues found that in human cells, ANG II, unlike what was observed in other species, induced the dose-dependent and strong stimulation of human proximal tubular transport via the activation of AT1-dependent nitric oxide/guanosine 3’, 5’-cyclic monophosphate/ERK [106]. Currently, the molecular mechanisms underlying species-specific differences in proximal tubular responses to the NO/cGMP pathway remain unknown [107].
The effects of ANG II on HCO3- transport in the thick ascending limb are controversial. ANG II inhibits HCO3- absorption via a cytochrome P-450–dependent mechanism [108] and stimulates NHE3 [101]. In addition, ANG II stimulates Na+/H+ exchange in the early distal tubule and amiloride-sensitive Na+ channels in the late distal tubule of cortical nephrons [104].
Studies using in vivo microperfusion have confirmed that a low dose of luminal ANG II stimulates Na+/H+ exchange in the early and late distal segments of the rat kidney in addition to vacuolar H+-ATPase in late distal segments [109]. In the rabbit cortical collecting duct, peritubular ANG II stimulated β cell apical HCO3- secretion through a basolateral AT1 receptor [105]. In intercalated cells of connecting tubules and cortical collecting ducts, ANG II stimulated H+-ATPase and may have contributed to the regulation of chloride reabsorption and bicarbonate secretion [110]. In the α intercalated cells of the mouse cortical collecting duct, ANG II stimulated the secretion of H+ into the lumen, which drove Cl- reabsorption via apical Cl-/HCO3- exchange and generated a more favorable electrochemical gradient for epithelial Na+ channel (ENaC)-mediated Na+ reabsorption [111]. ANG II stimulated ENaC in the rat cortical collecting duct via a Ca2+-independent PKC pathway that activated NADPH oxidase (NOX), resulting in an increase in superoxide generation. This stimulatory effect of ANG II on ENaC may be partially responsible for blocking the arachidonic acid-induced inhibition of ENaC [112].
Dose-dependent biphasic effects of ANG II on Na+/H+ in the distal nephron
In research performed using MDCK cells (a permanent cell line with morphological and physiological similarities to cells in the collecting duct), we demonstrated that ANG II has a dose-dependent biphasic effect on Na+/H+ in the distal nephron [113]. In this study, we used fluorescent probes to measure the recovery rate of intracellular pH [(pHi)r] after inducing the acidification of the pHi via an NH4Cl pulse, and we also monitored the [Ca2+]i. Figure 1 shows that in the controls, the (pHi)r was 0.088 ± 0.014 pH units/min [114]. The addition of ANG II (10-12 M) to the bath caused a significant increase (38%) in the (pHi)r, and in the presence of ANG II (10-9 M), this increase was even more significant (123%). However, the addition of ANG II (10-7 M) significantly decreased it (77%). Using Atrial Natriuretic Peptide (ANP) or dimethyl-BAPTA-AM alone did not affect the (pHi)r but impaired both the stimulatory and inhibitory effects of ANG II on (pHi)r. The (pHi)r also decreased significantly when EGTA was applied alone (73%) or in addition to ANG II (10-9 M) (71%), but was not significantly altered when EGTA was applied with ANG II (10-7 M). Figure 1 also shows that MDCK cells exhibited a mean baseline [Ca2+]i of 99 ± 7 nM [115]. The subsequent addition of ANG II (10-12, 10-9, and 10-7 M) increased [Ca2+]i progressively from control values to 130% in a dose-dependent manner. The addition of ANP (10-6 M) to the bathing solution led to a rapid and significant decrease in [Ca2+]i (70%), and the subsequent addition of ANG II resulted in the recovery of [Ca2+]I, which reached 50% without exceeding normal baseline values, even in the presence of ANG II (10-7 M). This figure also indicates that dimethyl-BAPTA-AM alone led to a significant decrease in [Ca2+]i (50%) and that the subsequent addition of ANG II resulted in the recovery of [Ca2+]i without achieving control values. However, the presence of EGTA in the cell suspension led to a significant decrease in [Ca2+]i (85%), and the subsequent addition of ANG II did not result in the recovery of [Ca2+]i. Hence, the results indicate that ANG II operates through different pathways (which lead to either a small or large increase in [C2+a]i) and has a dose-dependent bimodal effect on the regulation of (pHi)r by modulating Na+/H+ activity. ANP and dimethyl-BAPTA-AM cause a moderate decrease in [Ca2+]i that does not affect (pHi)r but does impair the pathway that causes an increase in [Ca2+]i, thereby blocking both the stimulatory and the inhibitory effects of ANG II on this process. In addition, EGTA caused a sharp reduction in [Ca2+]i, impaired (pHi)r and blocked both the stimulatory and the inhibitory effects of ANG II on (pHi)r. In this study, experiments that were performed on per meant filter supports indicated that the Na+/H+ exchanger, which is responsible for Na+-dependent (pHi)r, is located on the basolateral membrane. Taken together, these results suggest that [Ca2+]i plays a role in regulating (pHi)r via a process that is mediated by NHE1 exchanger and stimulated/impaired by ANG II. To better understand this bimodal effect of ANG II on NHE1 and to support previous findings [11,116], it was suggested that site A of this exchanger (the high-affinity site on CaM) works as an autoinhibitory domain, and a discrete increase in [Ca2+]i (in the presence of low concentrations of ANG II) may induce Ca2+/CaM binding to this region, thereby blocking the inhibitory interaction, resulting in the activation of NHE1 (Figure 6). On the other hand, it is possible that site B of the exchanger (the low-affinity site for CaM) binds to Ca2+/CaM only at high [Ca2+]i (in the presence of high concentrations of ANG II). Under these conditions, it inhibits NHE1 activity (Figure 6). To test this hypothesis, we used site-directed mutagenesis to provide some new information regarding how each Ca2+/CaM-binding site in the carboxy-terminus of NHE1 confers responsiveness to ANG II [117]. This study indicated that under control conditions, Ca2+/CaM binding sites do not function to maintain the basal activity of NHE1. However, in the presence of slightly increased [Ca2+]i that was induced by low ANG II concentrations, site A seems to be responsible for the stimulation of NHE1, whereas in the presence of a significant increase in [Ca2+]i, which was induced by high levels of ANG II, site B plays an important role in maintaining the basal activity of the Na+/H+ exchanger. Therefore, these results showed that both sites play important regulatory roles in the calcium-dependent modulation of NHE1 by ANG II.
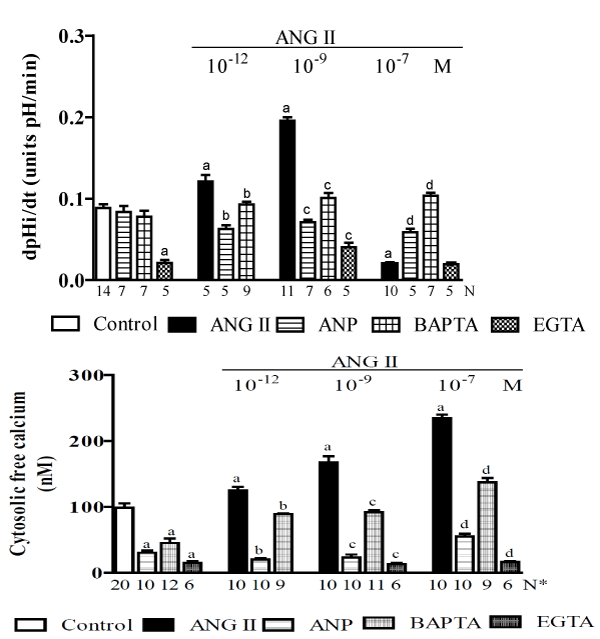
Figure 1. A study of MDCK cells [113] showing the effects of ANG II (at 10-12, 10-9 and 10-6M) alone or plus ANP (10-6 M), dimethyl-BAPTA/AM (5 X 10-5 M) or EGTA (2.5 mM) on the: A - pH intracellular recovery rate and B - cytosolic free calcium concentration. Values are shown as the means ± SE. N= no. of observations. N* = no. of experiments in which the maximum fluorescent signal was averaged for 10 cells. dpH/dt = pH intracellular recovery rate in the first 2 min after a NH4Cl pulse. a = P < 0.05 vs. control. b = P < 0.05 vs. ANG II (10-12 M). c = P < 0.05 vs. ANG II (10-9 M). d = P < 0.05 vs. ANG II (10-7 M).
In more recent studies, it was demonstrated that in MDCK cells, the regulation of NHE1 and NHE3 activity by ANG II is mediated by the activation of the ANG II type I receptor/phospholipase C/calcium/calmodulin pathway [118].
Over the past 15 years, our understanding of the RAS has substantially increased, and it is currently accepted that both circulating and tissue RAS are far more complex than previously thought. Thus, in addition to its traditional components (described in section 2.2: ANG II in the present review), the modern concept of the RAS includes the following: a new enzyme (ACE2) [119,120], peptides such as ANG-(1–7) and ANG A [121], the pro-renin receptor [122], Mas receptor [123], and Mas-related G-protein–coupled receptor D [124] and the heptapeptide alamandine [124]. However, although ANG III and ANG IV, the smaller peptide fragments of the RAS, also have biological activity, their plasma levels are much lower than the levels of ANG II or ANG-(1-7) [125,126].
This area of research has been described in several previous reviews [52,127-133].
The identification of the ACE homolog ACE2, an enzyme that is important for the generation of ANG-(1–7), and the G protein-coupled receptor Mas, which is encoded by the Mas proto-oncogene and is a receptor for ANG-(1–7), allowed researchers to determine that the RAS system contains at least two cascades: i) the ACE2-ANG-(1–7)-Mas axis, which probably act as the counterregulatory portion of ii) the classical RAS axis, or the ACE-ANGII-AT1 and AT2 receptors axis. The Mas gene is expressed in the brain, testes, kidney, heart [134-136] and central nervous system, where it is found in various regions, including cardiovascular regulatory areas [137]. In agreement with the activities that were previously described for ANG-(1–7), Mas-deficient mice exhibited increased blood pressure, impaired endothelial function, decreased NO production, and decreased endothelial NO synthase expression [138,139]. Also in agreement with findings showing the cardioprotective effects of ANG-(1–7), a genetic deletion of the Mas receptor impaired heart function and changed the extracellular matrix to a profibrotic state [140]. After the activation of the Mas receptor, the intracellular signal transduction mechanisms that are involved in the following processes or tissues are poorly understood: i) in vivo, in the rat heart, ANG-(1–7) stimulated the phosphorylation of Janus kinase 2 (JAK2), insulin receptor substrate (IRS)-1 and Akt [141], ii) Mas receptor activation led to an increase in NO production via the phosphorylation of eNOS, a process that involves the activation of phosphatidylinositol 3-kinase-dependent Akt phosphorylation [142,143], and iii) upon the activation of the Mas receptor, MAPK phosphorylation is inhibited [144,145].
ANG-(1–7) can also bind to the AT1 and AT2 receptors, although only at high hormonal concentrations [146-148]. However, studies indicating that AT2 is involved in ANG-(1-7)-induced vasorelaxation have not produced conclusive data [124,148-150], and the possibility of a physical or functional interaction between Mas and AT2 should be considered [152-155].
In addition, the existence of a new ANG-(1–7) receptor subtype has been suggested [156], and an interaction between ANG-(1–7) and different ANG II receptors has also been proposed [157,158]. In hypertensive animals, ANG-(1–7)–induced vasodilation was restored by acute or chronic AT1 blockade with losartan, suggesting an interaction between AT1 and Mas [157-159]. Furthermore, the contribution of AT2 and bradykinin B2 receptor (BKR) to the vascular effects of ANG-(1–7) should not be disregarded, and they suggest the potential presence of crosstalk between BKR with Mas and AT2 [148,160,161].
ANG-(1–7) is formed from ANG I and ANG II via the activity of ACE, ACE2, and several other enzymes [162]. ACE is the main enzyme that is responsible for the conversion of ANG I into ANG II, and ACE2 then cleaves ANG II into ANG-(1–7) [163]. Furthermore, ACE2 forms ANG-(1–9) from ANG I, and ANG-(1–9) can be converted into ANG-(1–7) by ACE [119]. However, the preferable physiological substrate for ACE2 is ANG II [120,163].
ANG-(1–7) is subsequently metabolized into an inactive fragment, ANG-(1–5), by ACE [164-166]. The half-life of ANG-(1–7) is several seconds, and ACE inhibitors, which inhibit the metabolism of ANG-(1–7) into ANG-(1–5), increase the half-life of ANG-(1–7) [167].
ACE2 is expressed in the heart, kidneys, and testes and, to a lesser extent, the liver, lungs, small intestines and brain [168-170]. In the kidney, ACE2 gene expression has been observed in glomeruli, the vasa recta and all nephron segments except for the thick ascending limb [171]. Additionally, relatively high amounts of ACE2 were detected in the apical brush-border membrane of proximal tubule epithelia, where it colocalized with ACE [171].
Actions of Ang-(1–7) in the kidney
Several studies have demonstrated that the kidney is an additional target of ANG-(1–7) activity. However, the renal effects of this heptapeptide are incompletely understood. In isolated, perfused rat kidneys and anesthetized animals [172,173], ANG-(1–7) has been shown to increase sodium and water excretion and the glomerular filtration rate (GFR) without affecting renovascular resistance. Hence, it is also possible that ANG-(1–7) induces diuresis and natriuresis by inhibiting renal tubular Na+/K+-ATPase [174,175]. However, ANG-(1–7) also induced antidiuresis in water-loaded rodents (258) and increased renal tubular sodium reabsorption in rats [176]. In addition, in normal and hypertensive rats [177] and in water-loaded [178] or virgin female rats [179], ANG-(1–7) had an antidiuretic effect that was mediated, at least in part, by the Mas receptor. Furthermore, in Sprague-Dawley rats, the administration of ANG-(1–7) resulted in antidiuresis that was associated with an upregulation in renal aquaporin-1 [180], and in isolated rat inner medullary collecting duct cell suspensions, ANG-(1–7) (10−9 M) increased cAMP production [181], a response that was attenuated by Mas receptor antagonists and the pharmacological blockade of the AVP V2 receptor. ANG-(1–7) also caused afferent arteriolar vasodilation and antagonized the vasoconstrictor effects of ANG II [114].
Actions in the proximal tubule: In the proximal tubular segment, the actions of ANG-(1-7) are often conflicting. In the proximal tubule of anesthetized animals, ANG-(1–7) increases urinary flow rates and sodium excretion. This effect was abolished by the Mas receptor antagonist A779 in rats [175], but in dogs, this hormonal effect was partially blocked by the AT1 receptor antagonist EXP 3174 but not by the AT2 receptor antagonist PD123319 [182]. In isolated rat proximal straight tubules, similar to ANG II, ANG-(1–7) exerts a biphasic effect through AT1 receptors. At physiological levels (10−12 M), ANG-(1–7) increases fluid and bicarbonate reabsorption, while at high concentrations (10−8 M), ANG-(1–7) decreases fluid reabsorption [183]. In rabbit proximal tubular cells, ANG-(1–7) inhibits sodium reabsorption by activating phospholipase A2 [184]. In addition, in isolated basolateral membranes of kidney proximal tubules [175,185], ANG-(1–7) modulated the activity of Na+/K+-ATPase, and in isolated pig kidney inner cortical membranes [186], it inhibited Na+-ATPase activity via an effect that was reversed by an AT2 receptor antagonist. Furthermore, in isolated proximal tubules, i) ANG-(1–7) stimulated the release of arachidonic acid [184], and ii) the inhibition of prostaglandin release that resulted from COX inhibition attenuated the ANG-(1–7)-induced increase in urine flow and sodium excretion [187]. Taken together, these results suggest that in the proximal tubule, ANG-(1–7) affects natriuresis and diuresis by activating Mas receptors. However, AT1 and AT2 receptors may also be involved.
Dose-dependent biphasic effect of ANG-(1–7) on proximal Na+/H+ exchanger: Because i) the nature of the mechanism underlying the effect of ANG-(1–7) on proximal nephron bicarbonate reabsorption has not yet been clearly defined [175,182-184], ii) apical NHE3 mediates most NaCl, NaHCO3−, and fluid reabsorption by the renal proximal tubule and is critical for the normal maintenance of extracellular fluid volumes, blood pressure, and acid-base balance [188], and iii) under physiological conditions, plasma concentrations of ANG-(1–7) are in the picomolar range but can increase under conditions involving extracellular volume expansion [189,190], the purpose of a recent study by our laboratory [191] was to determine the acute direct effects of different doses of ANG-(1–7) on net bicarbonate reabsorption (JHCO3-) via the NHE3 exchanger in vivo in the proximal convoluted tubules (S2 segment) of normal Wistar rats. The results were evaluated using stationary microperfusions via H ion-sensitive microelectrodes. Figure 2 shows that the control JHCO3- was 2.82 ± 0.078 [192] nmol/cm-2.s-1. ANG-(1–7), at a low hormonal dose (10−12 or 10−9 M), significantly inhibited (37% or 61%, respectively) while a high hormonal dose (10−6 M) stimulated (90%) the NHE3 exchanger. This biphasic action of ANG-(1–7) on Na+/H+ was the reverse of the biphasic action that was described for ANG II, i.e., as previously discussed, it is widely accepted that ANG II stimulates Na+/H+ exchange at low doses and inhibits it at high doses (Figure 1). Figure 2 also shows that dimethyl-BAPTA-AM alone did not affect the JHCO3- concentration but did impair both the inhibitory and the stimulatory effects of ANG-(1-7) on it. In addition, Thapsigargin alone inhibited JHCO3- and impaired both the inhibitory and stimulatory effects of ANG-(1-7) on it. Figure 2 also shows that the proximal convoluted tubule exhibited a mean baseline [Ca2+]i of 101 ± 2 nM [164]. The addition of ANG-(1-7) (10-12, 10-9, and 10-6 M) increased [Ca2+]i (by 124%, 100% and 78%, respectively). The addition of BAPTA led to a significant decrease in [Ca2+]i (by 42%), and the addition of ANG-(1-7) resulted in a recovery of [Ca2+]i without achieving baseline values. Thapsigargin alone or with ANG-(1-7) (10-9 or 10-6 M) increased [Ca2+]i (by approximately 350%). The data in that study also showed that the biphasic effect of ANG-(1–7) on the Na+/H+ exchanger occurred via the Mas receptor and that A779 (a Mas receptor antagonist) abolished this hormonal biphasic effect. These results are also consistent with previously described findings in the literature that indicated that the renal effects of ANG-(1–7) are dose dependent and mediated, at least in part, by the Mas receptor [193-196]. Nevertheless, in isolated rat proximal straight tubules (S3 segment), it was shown that, like ANG II, ANG-(1–7) exerts a biphasic effect through AT1 receptors: at physiological levels, ANG-(1-7) increases fluid and bicarbonate reabsorption, while at high concentrations, ANG-(1-7) decreases these parameters [183]. This finding is in contrast to previous results described from in vivo experiments involving rat proximal convoluted tubules (S2 segment) [191]. Thus, whether one or more different signaling systems are involved in mediating the effect of ANG-(1–7) on NHE3 activity in the S2 and S3 proximal segments remains to be established. In addition, the renal effects of ANG-(1–7) may also involve AT1 and AT2 receptors [182,184,186] and V2 receptors [181], indicating the existence of a cross-talk mechanism between the Mas receptor and the ANG II and AVP systems. Furthermore, in a study of isolated rat proximal straight tubules [183], the actions of ANG-(1–7) were blocked by an AT1 antagonist, suggesting a different site of action than the one that was assessed in the in vivo rat proximal convoluted tubules study [191]. However, the results of the in vivo rat proximal convoluted tubules study were consistent with the hypothesis presented in the current literature [128,197], which suggests that both ANG-(1–7) and ANG II have opposite effects on several cardiovascular mechanisms and that at physiological doses, ANG-(1–7) is a vasodilator and ANG II is a vasoconstrictor. In addition, the results of in vivo experiments on rat proximal convoluted tubules [191] were compatible with the notion that the NHE3 exchanger is stimulated by a moderate increase in [Ca2+]i in the presence of a high dose of ANG-(1–7) (10−6 M) and that the inhibition of this exchanger largely increases the [Ca2+]i that was induced by low doses of ANG-(1–7) (10−12 or 10−9 M). These findings are in opposition to those described in previous studies of ANG II [113,198]. Additionally, the experiments in the in vivo rat proximal convoluted tubules study used BAPTA or thapsigargin [191] and confirmed the role of cytosolic calcium in the regulation of NHE3. Thus, the interaction between the opposing dose-dependent effects of ANG-(1–7) and ANG II on the renal proximal Na+/H+ exchanger and [Ca2+]i levels may represent an important physiological regulatory mechanism that controls extracellular volume and/or changes in pH. However, this is a complex mechanism, and additional factors need to be investigated. In addition, other factors must be considered. For example, in the in vivo rat proximal convoluted tubules study [191], the effects of luminal perfusions of ANG-(1–7) were examined using a stopped-flow microperfusion technique that is not affected by glomerular filtration rates and allows the researcher to measure tubular acidification on line [72]. Moreover, the experimental conditions were such that the measurements were performed after pre-determined amounts of hormone were added to the in vivo tubules. Therefore, the hormonal effects were local and did not cause systemic hemodynamic changes because the amount of hormone that was introduced into the tubules was insignificant. This result was confirmed by the observation that there were no changes in urine flow and osmolality, Na+ excretion, or systemic acid-base values during the experiments in which luminal hormonal microperfusions were performed [191]. Furthermore, in this study, the luminal perfusion solution for ANG-(1-7) contained raffinose (a nonresorbable molecule), which was added to achieve isotonicity. This prevented fluid reabsorption from being induced by the hormone. Thus, it is possible that the conflicting and incompletely understood proximal effects of this heptapeptide that are described in the current literature can be explained by the fact that in several studies in which ANG-(1–7) was injected parenterally, induced systemic hemodynamic effects under conditions in which the measurements were not made at determined hormonal levels within the proximal tubule.
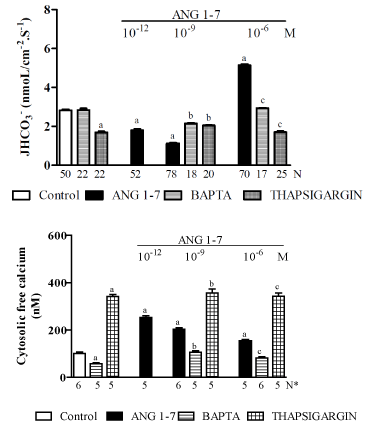
Figure 2. An in vivo study of the proximal convoluted tubules of normal Wistar rats [191] showing the effects of ANG-(1-7) (10-12, 10-9 or 10-6 M) alone or plus dimethyl-BAPTA/AM (5 X 10-5 M) or Thapsigargin (10-5 M) on: A - net bicarbonate reabsorption and B – the cytosolic free calcium concentration. Values are shown as the means ± SE. N= no. of observations. N* = no. of experiments in which the maximum fluorescent signal was averaged for 10 cells. JHCO3− = net bicarbonate reabsorption. a = P < 0.05 vs. control. b = P < 0.05 vs. ANG-(1-7) (10-9 M). c = P < 0.05 vs. ANG-(1-7) (10-6 M).
Given that i) it is widely recognized that the RAS plays a role in the pathophysiology of cardiovascular and renal diseases and that under such situations, the beneficial effects of ANG-(1-7) are in opposition to the deleterious effects of ANG II [199], and ii) since the plasma concentrations of ANG-(1–7) are higher in hypertensive rats [126,189,190], it is likely that in this situation, the high plasma levels of ANG-(1-7) have an inhibitory effect on NHE3 in the proximal tubule, which should ease hypertension. Hence, the purpose of a current study in our laboratory is to clarify the direct effects of ANG-(1–7) on the NHE3 exchanger in vivo in the proximal convoluted tubules of SHR rats (hypertensives) and their controls, WKY rats (normotensives) [unpublished data]. Figure 3 shows that in WKY rats, the control proximal JHCO3- that is maintained by the NHE3 exchanger is 2.41 ± 0.12 (57) nmol/cm-2s-1. ANG-(1–7), when applied at a low dose (10−9 M), significantly inhibited (by approximately 30%) while a high dose (10−6 M) stimulated (by approximately 70%) the activity of the NHE3 exchanger. This biphasic action of ANG-(1–7) on NHE3 in the WKY rats was similar to the biphasic action that was described for ANG-(1-7) in Wistar rats (see Figure 2). Figure 3 also shows that the proximal convoluted tubule in the WKY rats exhibited a mean baseline [Ca2+]i of 99 ± 3 nM [193]. The subsequent addition of ANG-(1-7) (10-9 or 10-6 M) increased the [Ca2+]i i (by approximately 100% and 58%, respectively). However, in the SHR rats, the control proximal JHCO3- was 1.98 ± 0.13 [200] nmol/cm-2s-1, and applying ANG-(1–7) at a low dose (10−9 M) significantly stimulated (by 57%) while a high dose (10−6 M) significantly inhibited (by 38%) the NHE3 exchanger. Hence, the biphasic action of ANG-(1–7) on NHE3 in the SHR rats was similar to the effect observed for ANG II in Wistar rats (Figure 1). Therefore, these results indicate that in hypertensive animals, a high plasma concentration of ANG-(1-7) [126,189,190] attenuates hypertension. Figure 3 shows that proximal convoluted tubules in SHR rats exhibited a mean baseline [Ca2+]i of 96 ± 2 nM [195], and the subsequent addition of ANG-(1-7) (10-9 or 10-6 M) increased it (by approximately 30% and 63%, respectively). The biphasic effects of ANG-(1-7) on the Na+/H+ exchanger in Wistar, SHR (hypertensives) and controls WKY (normotensives) rats are summarized in Figure 6.
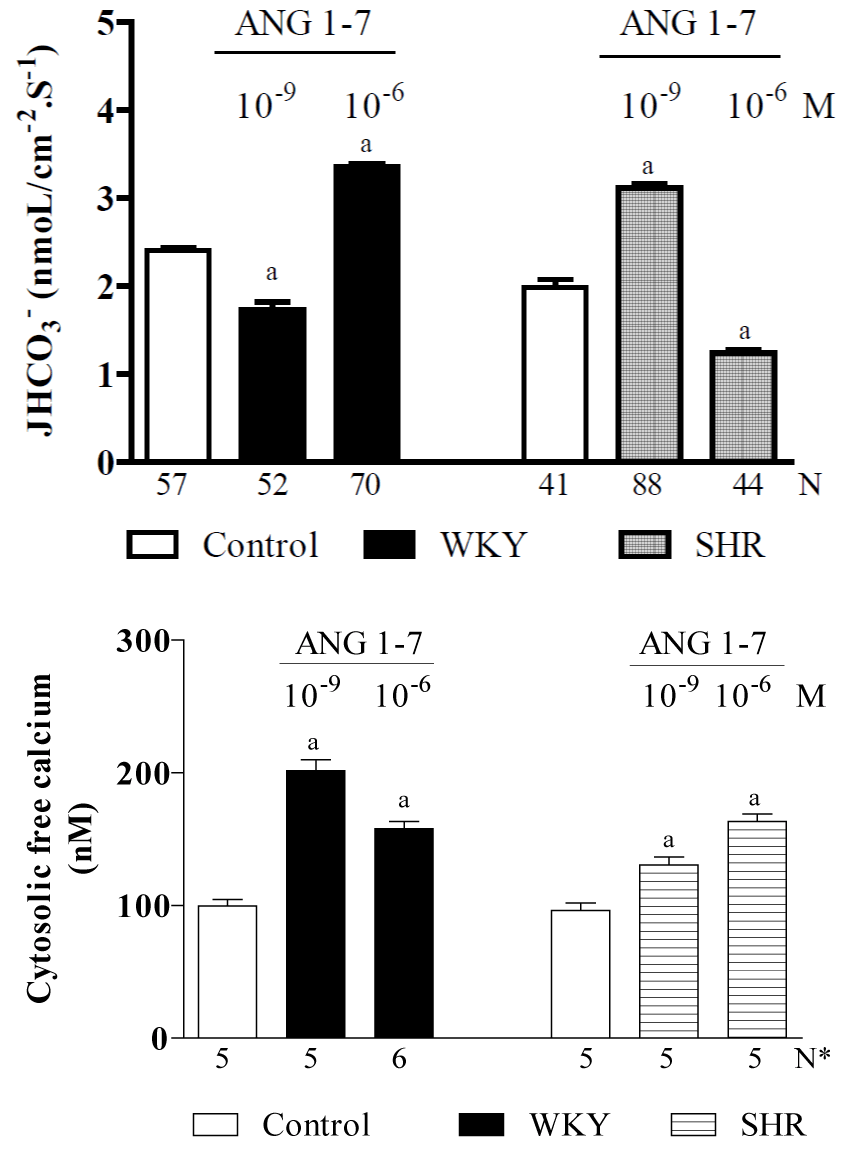
Figure 3. Current studies of in vivo proximal convoluted tubules in SHR (hypertensive) rats and their WKY (normotensive) controls, indicating the effects of ANG-(1-7) (10-9 or 10-6 M) on: A - net bicarbonate reabsorption and B – cytosolic free calcium concentrations [unpublished data]. Values are shown as the means ± SE. N = no. of observations. N* = no. of experiments in which the maximum fluorescent signal was averaged for 10 cells. JHCO3− = net bicarbonate reabsorption. a = P < 0.05 vs. control.
ALDO is a steroid hormone with mineralocorticoid activity that plays an important role in the maintenance of Na+, K+, water and acid-base balance via effects on renal electrolyte excretion. ALDO is produced mainly by the adrenal glomerulosa, but there is also evidence showing that it can be synthesized in other tissues. The most convincing evidence has been found in the central nervous system, whereas data suggesting that this hormone is produced in the heart remain controversial. The evidence demonstrating the extra-adrenal secretion of ALDO, coupled with its known non-epithelial actions, has led to speculation that ALDO may act in such tissues in an autocrine or paracrine manner [201,202].
Adrenal-derived ALDO is the principal source of the circulating and locally available supply of this hormone. Several factors regulate ALDO production, including adrenaline, vasoactive intestinal polypeptide, serotonin, ouabain, atrial natriuretic peptide, dopamine, heparin and adrenomedullin. Recently, novel factors that are secreted by adipose tissue have also been shown to stimulate ALDO synthesis in vitro. However, the principal regulators of the synthesis and secretion of this hormone are ANG II, the concentration of extracellular potassium and ACTH [203,204].
It was classically accepted that ALDO is synthesized in the adrenal zona glomerulosa and that its lipophilic nature causes it to enter the cell via diffusion through the cellular membrane. It then binds to a specific mineralocorticoid receptor (MR) that is located in the cytosol of target cells in epithelial and non-epithelial tissues. The translocation of the resulting steroid receptor complex to the cell nucleus modulates the gene expression and translation of specific ALDO-induced proteins that regulate electrolyte and fluid balance, and consequently blood-pressure homoeostasis. ALDO may also act through alternative receptors in epithelial and non-epithelial tissues in a rapid non-genomic manner that is independent of gene transcription and translation. The modulatory effect of non-genomic signalling responses on the transcriptional effects of MR is an emerging theme, and the relevance of these events to the effects of ALDO on whole-body electrolyte and acid-base homeostasis remains to be determined. In addition, for more than a decade, it has been accepted that an increase in ALDO circulating levels, in combination with a high-sodium diet, induces hypertension and cardiovascular and renal damage that are triggered by, among other factors, MR, epidermal growth factor receptor (EGFR) and oxidative stress independent of the RAS [205].
Regulation of ALDO biosynthesis
Renin–Angiotensin system: ALDO biosynthesis is principally regulated by the RAS, a system that is described above in sections 2.2 ANG II and 2.3 ANG-(1-7) of the present review. The stimulation of Renin release increases the plasma levels of ANG II, which subsequently stimulates the secretion of ALDO from the adrenal glands [206]. The stimulation of the formation of ANG II and ALDO increases sodium reabsorption and consequently increases the sodium content of the body, which, in turn, inhibits Renin gene expression [206-208]. Hence, the in vivo effect of ALDO may be an indirect consequence of changes in the sodium content of the body, extracellular volume and/or blood pressure. It is also possible that ALDO exerts a direct positive effect on Renin gene expression at the cellular level, probably by stabilizing Renin mRNA [52]. The adrenal response to ANG II occurs within minutes, a time course that implies that no new protein synthesis is required. The acute, ANG II-mediated release of ALDO may involve its rapid synthesis from intermediate compounds in the steroid genic pathway or its de novo synthesis from cholesterol, possibly as a consequence of Star protein activation, which leads to an increase in the transport of cholesterol to the inner mitochondrial membrane.
Extracellular K+ concentration ([K+]e): The production of ALDO is acutely sensitive to very small changes in [K+]e. Increased [K+]e stimulates ALDO secretion, which helps to maintain K+ homoeostasis. The effects of [K+]e and ANG II are synergistic, so that the prevailing [K+]e determines the concentration/effect of ANG II-mediated ALDO production [203,204,206].
ACTH: Despite the opposing effects that have been observed in the presence of acute and chronic ACTH, there is no doubt that this hormone is involved in the normal, physiological regulation of ALDO production [209-211].
Intracellular pathways of ALDO
Important intracellular signalling peptides that are involved in ALDO activities include epidermal growth factor (EGF) and its receptor (EGFR) [212], which participate in signalling events related to the activation of G protein-coupled receptor proteins, growth hormones and cytokines via a transactivation mechanism that may play a central role in signal transduction [213]. The interaction between MR and ALDO triggers genomic actions and also promotes the transactivation of the EGFR, an event that is necessary for the non genomic activation of the ERK1/2 cascade. These are mitogen-activated protein kinases (MAPKs), which are a group of serine/threonine kinases that, when stimulated, phosphorylate their substrates at serine and/or threonine residues. These kinases are involved in transducing signals from the cell membrane to the nucleus [214]. The ERK1/2 signalling pathway is activated by RAF-type MAP kinases. When these kinases are phosphorylated, they activate the MAP kinases MEK1 and MEK2, which themselves phosphorylate and activate the ERK1/2 kinases. When activated, the ERK1/2 kinases phosphorylate several effector molecules that are involved in many fundamental cellular processes, such as differentiation, proliferation, apoptosis, survival and metabolism [215]. In the kidneys, the ERK1/2 pathway participates in the hormonal regulation of different ionic carriers along the nephron, and they play an important role in maintaining the homeostasis of extracellular fluids [216].
In addition to the important physiological role of ALDO in the kidney, in recent years, the role of ALDO in the pathogenesis of renal injury has also been extensively studied, and several studies have indicated that ALDO and its classic receptor (MR) are involved in the development of inflammation, oxidative stress, glomerulosclerosis, fibrosis, and apoptosis in renal tissue [217-221]. Additionally, in vivo and in vitro studies have suggested that ALDO contributes to the occurrence of apoptosis in glomerular, tubular and mesangial cells [219,222,223]. According Briet and Schiffrin [224] and Brown [200], the signaling pathways that potentially involve ALDO in the induction of inflammation, oxidative stress, apoptosis and fibrosis in the kidney could include the following. i) After entering the cell, ALDO binds to cytoplasmic receptors (MR and/or GR) to promote the transactivation of EGFR and the phosphorylation of c-Src, resulting in the phosphorylation of ERK1/2 and pro-fibrotic responses. In addition, C-Src also stimulates the function of NADPH oxidase (probably NOX 4) and the production of reactive oxygen species (ROS), which participate in the induction of inflammation, oxidative stress, apoptosis and fibrosis in kidney tissues. ii) ALDO binds to a membrane receptor (GPR30) to stimulate the transactivation of EGFR and the phosphorylation of ERK1/2 by triggering the same processes described above. iii) Finally, the translocation of a complex containing MR and/or GR-ALDO into the nucleus promotes the transcription of fibrosis-related (e.g., PAI-1 and TGFb-1) and apoptosis (e.g., BAX and BCL-2) genes.
Classic genomic actions of ALDO
Epithelial actions: Classically, it has been accepted that ALDO acts on epithelial cells, particularly in the renal collecting duct, where it regulates the transport of Na+, K+, H+ and water. The main functions of classic genomic ALDO activity in epithelial cells are: i) Na+ reabsorption across the apical membrane, which is mediated by the luminal amiloride-sensitive epithelial Na+ channel (ENaC); ii) transport across the basolateral membrane, which is driven by the ouabain-sensitive Na+/K+- ATPase; iii) the secretion of K+ from the cell into the lumen through luminal K+ channels; iv) the secretion of H+ through the luminal vacuolar H+-ATPase; and v) water transport, which follows the movement of Na+ across the membrane. Apical Na+ channel activity is the limiting step in the transport process, and it is likely that ALDO ultimately acts to increase the open time of existing ion channels and/or increase the total number of such channels [225]. However, other protein targets have also been identified, including i) a luminal NHE3 exchanger in the colon [226,227] and the proximal tubule [228] and a basolateral NHE1 in the proximal tubule [229,230], ii) an Na+/K+-ATPase in human kidney proximal tubule (HKC11) cells [231], iii) a luminal thiazide-sensitive Na+/Cl− cotransporter in the distal renal tubule that appears to mediate Na+ reabsorption in response to volume depletion [232], and iv) a renal proximal H+-ATPase [233].
The genomic epithelial actions of ALDO operate in early (1 – 6 h) and late (> 6 h) phases. The genomic early phase is mediated by changes in gene expression that activate ion channels and signaling proteins, which then induce electrolyte-transport proteins. The genomic late phase results from both primary and secondary effects on gene expression. One of the early ALDO-induced proteins is Sgk1, a serine-threonine kinase [234,235]. Presumably, Sgk1 binds and phosphorylates the ENaC regulatory protein (Nedd4–2) to reduce its binding to ENaC [236]. A subsequent reduction in ENaC ubiquitination by Nedd4–2 increases ENaC density and stability at the apical membrane, resulting in increased ENaC-dependent Na+ reabsorption. In addition, during the early phase of genomic ALDO action, the expression of the small, monomeric Kirsten Ras GTP-binding protein-2A (Ki-RasA) is induced and required for the ALDO-mediated effects on Na+ transport in renal epithelial cells. Ki-RasA appears to have dual contrasting effects on ENaC channels by i) keeping the channel open and ii) decreasing the number of channels in the plasma membrane [235,237,238]. The activity of the lipid kinase PI3K is increased by ALDO in the kidney [235,239], and the inhibition of PI3K reduces both the early and late genomic actions of ALDO [225].
The corticosteroid hormone-induced factor (CHIF) is expressed in the basolateral membranes of epithelial cells in the distal colon and nephron [235,240]. ALDO probably stimulates CHIF expression, and the resulting protein interacts with final effectors to promote ion transport [241,242].
Target tissue specificity: Although it has been almost 60 years since ALDO was first isolated and its physiological effects on renal function identified [243], until the end of the last century, the specificity of ALDO action was believed to be conferred by the abundant presence of high-affinity type 1 MR in the cytosol of ALDO target tissues [244]. This receptor belongs to the nuclear receptor superfamily and is composed of several functional domains that include an N-terminal domain, a highly conserved DNA-binding domain and a C-terminal ligand-binding domain [244]. ALDO binding results in a conformational change and a dissociation of associated proteins followed by dimerization and translocation to the cell nucleus [245] and the subsequent activation or repression of transcriptional activity [246].These effects can be inhibited by actinomycin and cycloheximide, which block transcription and translation, respectively [225].
The MR is distinguishable from the glucocorticoid receptor (GR), which is ubiquitously expressed and exhibits a higher affinity for glucocorticoids. The lack of specificity of the MR and the fact that plasma levels of glucocorticoids are 1000 times higher than those of ALDO suggest that MR should be predominantly occupied by glucocorticoids. Hence, the ability of ALDO to act on MR was partly explained by the fact that the majority of glucocorticoids are bound to proteins in plasma. However, the concentration of free glucocorticoids in plasma is about 100 times higher than the concentration of ALDO, suggesting that there must be an additional mechanism underlying the action of ALDO in the target tissues independent of the MR [225].
Non-epithelial actions: MR is also localized in a number of non-epithelial tissues, especially in the cardiovascular system (CVS) and central nervous system (CNS). However, while the properties of the MR in these tissues are largely similar, the effects that MR mediates are extremely diverse. In the CVS, ALDO promotes cardiac hypertrophy, fibrosis and abnormal vascular endothelial function, while in the CNS, it appears to regulate blood pressure, salt appetite and sympathetic tone [247-250].
Crosstalk between ALDO and ANG II also plays a role in regulating the migration of vascular smooth muscle cells via interactions with c-Src-regulated redox-sensitive RhoA pathways [251], which emphasizes the benefits of dual therapy [252]. In addition, the subcommissural organ is involved in the central regulation of ALDO secretion and sodium homoeostasis, while an amygdala MR has been implicated in the control of salt appetite [253].
Non-genomic actions of ALDO
Since the end of the last century, in addition to its classic genomic activities, ALDO has been demonstrated to induce rapid cellular non-genomic effects. On the basis of several lines of evidence [254-257], it has been proposed that non-genomic ALDO actions i) are mediated by a distinct and specific membrane receptor that is different from the classical intracellular MR, ii) are insensitive to classical MR antagonists, such as canrenone or spironolactone, iii) are based on a high affinity for ALDO but not for glucocorticoids. The physiological and clinical relevance supporting these rapid effects remains unclear, but their existence has been described in various target organs and cells, including amphibian skin and urinary bladders [258,259], vascular smooth muscle cells and endothelial cells [260], skeletal muscle cells [261], human mononuclear leukocytes [262], cardiac myocytes [263], skin fibroblasts from MR-knockout mice [256], colonic epithelial cells [264] and isolated colonic crypts [265]. Several sites in the kidney, particularly cultured kidney cells, have been shown to be sensitive to non-genomic ALDO action [266], including principal cells that were freshly isolated from rabbits [267], the human distal colon [268], in vivo renal proximal tubules (S2 segment) [228], isolated renal proximal tubules (S3 segment) [229,233], medullary thick ascending limbs [269] and renal collecting duct cells [270]. Its non-genomic actions include effects on signal transduction pathways and ion transporters, such as the epithelial Na+ channel [267], the Na+/H+ exchanger [228,229,271,272] and the vacuolar H+-ATPase [230,233,258,259,273].
While the non-genomic actions of ALDO influence electrolyte homeostasis, pH and cell volume in classical MR target organs, these activities also contribute to pathophysiological effects in the renocardiovascular system that cause endothelial dysfunction, inflammation and remodeling [274]. However, the molecular mechanisms underlying the nongenomic actions of ALDO on electrolyte transporters and signaling enzymes and the consequences of these nongenomic actions on whole-body acid-base balance remain unknown [275].
The non-genomic receptor: The nature of the receptor that initiates rapid non-genomic ALDO-induced signaling remains unknown. However, evidence supporting its existence comes from several sources. Data in the literature indicate the following: i) the initiation involves MR in most instances or ii) the existence of a plasma membrane-binding site for ALDO. Although the presence of a high-affinity membrane-associated receptor that is insensitive to MR antagonism and is unable to bind glucocorticoids has been detected in the vascular endothelium [276], a full structural characterization of this non-genomic receptor has yet to be achieved. Some studies have indicated that rapid non-genomic responses are mediated by a variety of receptor types that are associated with the plasma membrane or its caveolae components, and these receptors may include a membrane-associated nuclear receptor [277]. The intermediate steps that couple the ALDO-MR interaction with the nongenomic activation of specific protein kinases are not fully characterized; however, the transactivation of EGFR is a crucial step in transducing this activating signal to various downstream signaling intermediates that are responsive to ALDO [278]. In addition, rapid ALDO effects also involve a multitude of signaling molecules and include cross-talk with genomic ALDO effects as well as with the ANG II receptor and EGFR [274]. More recent results in studies of human and mouse endothelial cells have indicated that striating (a calmodulin-binding protein) is a mediator for nongenomic mechanisms of ALDO and estrogen effects on pERK and phosphorylated eNOS, respectively, suggesting a unique level of interaction between the ALDO receptor and the estrogen receptor in the cardiovascular system [279]. Finally, it has been shown that the functions of non-genomic ALDO effects may be to modulate other signalling cascades, with mechanisms depending on the surrounding milieu [274].
Additionally, it has been shown that through both genomic and non-genomic mechanisms, ALDO stimulates Na+ reabsorption, K+ secretion [280], Na+/H+ exchange [230] and H+-ATPase [233] in the kidney, and Na+/H+ exchange in vascular smooth muscle cells [281].
Dose-dependent biphasic effect of ALDO on Na+/H+ exchanger
Approximately one decade ago, it was shown that i) ALDO rapidly increases Na+/H+ exchanger activity in a variety of cells, including distal colon and renal epithelial cells [255,282], and ii) ALDO also rapidly inhibits the Na+/H+ exchanger and HCO3- reabsorption in medullary thick ascending limbs [283]. Hence, the activation/inhibition of Na+/H+ exchanger by ALDO needed to be more clearly defined, because it was possibly that similar to ANG II [83,113,198], ALDO had a dose-dependent biphasic effect on the Na+/H+ exchanger (with low doses stimulating and high doses inhibiting the exchanger). In addition, it was also shown that an elevation in [Ca2+]i has the following effects: i) it serves as a second messenger in the ALDO-mediated initiation of nongenomic Na+/H+ exchanger activation [271], and ii) it is a pre-requisite for the genomic action of ALDO, and strong evidence indicates that pregenomic hormonal responses can influence genomic processes [284]. Therefore, we performed a study to clarify the genomic and nongenomic effects of ALDO on the Na+/H+ exchanger and the role of [Ca2+]i in these processes [230]. The experiments were performed using isolated rat proximal straight tubules (S3 segments) and fluorescent probes to measure pH intracellular recovery rates (pHi)r after the acidification of the pHi was induced by a NH4Cl pulse. We then monitored the [Ca2+]i. Figure 4 shows that in the controls, the (pHi)r was 0.191 ± 0.006 [137] units pH/min and that administering ALDO at 10−12 M after 2 min, 15 min or 1 h of preincubation (pi) resulted in an increase in the (pHi)r (by approximately 50%, 25% and 70%, respectively). However, administering ALDO at 10−6 M after 2 min, 15 min or 1 h pi significantly decreased the (pHi)r (by approximately 25%, 48% or 38%, respectively). ALDO, when administered at 10−12 M or 10−6 M, at 2 min, 15 min or 1h pi therefore had a dose-dependent stimulatory effect on [Ca2+]i. Results that are not shown indicated that at ∼1 min after the addition of ALDO (at either 10−12 or 10−6 M) to the bath, there was a transient and dose-dependent increase in the [Ca2+]i. At 15 min after the addition of ALDO, the [Ca2+]i was significantly higher, mainly for ALDO at 10−6 M. After 1 h of hormone addition, the [Ca2+]i remained high and was not different from the values found after 15 min. These results also indicated that adding actinomycin D (an inhibitor of gene transcription) or cycloheximide (an inhibitor of protein synthesis) did not influence the effects of ALDO (administered at either 15 or 2 min pi) but did inhibit the effects of ALDO (1h pi) on the (pHi)r and the [Ca2+]i (data not shown). Hence, this study indicated that ALDO had a dose-dependent biphasic effect on the exchanger that was mediated by nongenomic (15 or 2 min pi) and genomic (1 h pi) pathways. These data were compatible with results showing that the Na+/H+ exchanger was stimulated when cellular calcium was increased by a lower range of ALDO (at 10-12 M) and inhibited when cellular calcium was increased by high levels of ALDO (at 10-6 M). Hence, this biphasic action of ALDO on Na+/H+ is similar to the biphasic action described for ANG II (Figure 1). The genomic ALDO-induced regulation of the exchanger seems to be a mineralocorticoid-specific effect because the ALDO receptor antagonist spironolactone significantly inhibited the activity of the exchanger (results not shown). In addition, our data show that in the presence of HOE 694 (a specific inhibitor of basolateral NHE1), administered alone or with ALDO (10−12 or 10−6 M, 2 min pi), the (pHi)r was completely inhibited. However, in the presence of S3226 (a specific inhibitor of apical NHE3) alone, the (pHi)r was not different from control values, and S3226 also failed to prevent both the stimulatory effect of ALDO (10−12 M, 2 min pi) or the inhibitory effect of ALDO (10−6 M, 2 min pi) on the (pHi)r. Hence, these results indicated, for the first time, that ALDO modulates the mechanism regulating pHi via the NHE1 exchanger in the proximal tubule. The results of this research also indicated that MR and probably GR participate in the genomic and nongenomic effects of ALDO on [Ca2+]i and on the Na+/H+ exchanger. These data are in agreement with previous findings using RT-PCR that indicated the presence of these receptors in proximal S3 segments [230].
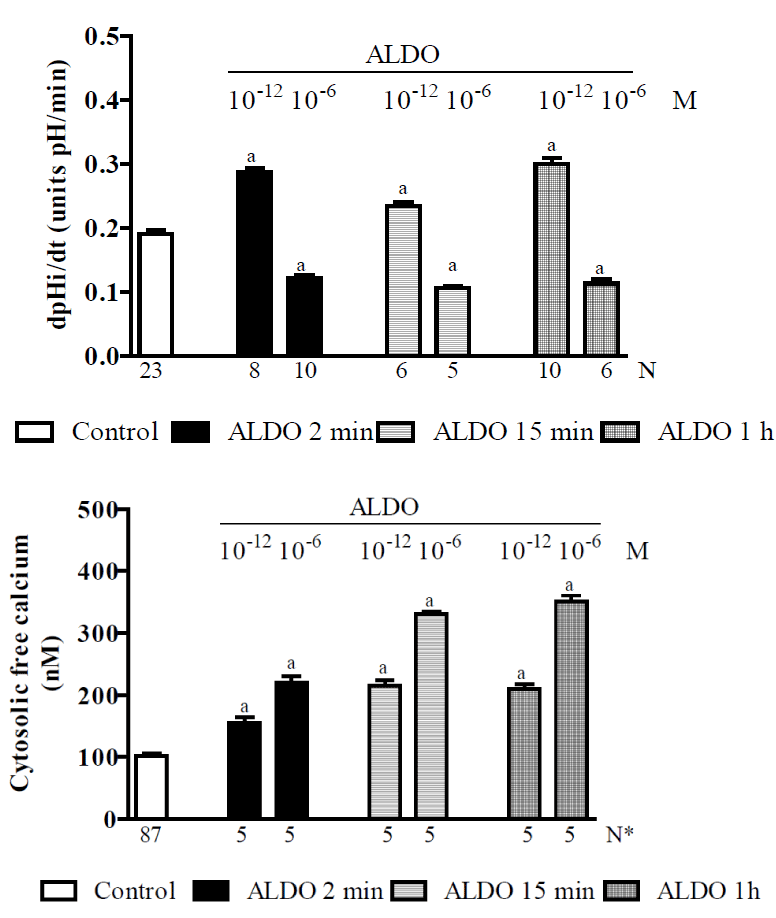
Figure 4. Research using isolated proximal S3 segments from normal Wistar rats [230] showing the effects of ALDO (10-12 or 10-6 M) after 2 min, 15 min or 1 h of preincubation on: A - pH intracellular recovery rates and B - cytosolic free calcium concentrations. Values are shown as the means ± SE. N = no. of observations. N* = no. of experiments in which the maximum fluorescent signal was averaged for 10 cells. dpH/dt = pH intracellular recovery rate in the first 2 min after a NH4Cl pulse. Pi = preincubation. a = P < 0.05 vs. control.
The Arginine vasopressin (AVP), or human antidiuretic hormone (ADH), is a neurohypophyseal cyclic octapeptide (1,099 D) with a 3-amino acid tail that plays an important role in water homeostasis and vasoconstriction and has functions in the kidney that have been well documented and will be the focus of this section of this Review. However, it has widely been recognized that AVP also has various nonpressor and nonantidiuretic activities that were reviewed in detail in a recent study [285]. The main site of AVP production is the hypothalamus. However, smaller amounts are also produced locally by many others tissues. Thus, AVP has endocrine, autocrine and paracrine effects. In normally hydrated persons, circulating AVP levels are very low (~ 1 pg/ml) because it is rapidly metabolized by the liver and excreted by the kidneys [286]. Hence, its half-life is 15 to 20 min. Because AVP is a small peptide, it is easily filtered through the glomerulus, but it is not metabolized in the kidney but is instead excreted in an unchanged form in the urine. Under normal physiological conditions, the very low concentration of AVP in plasma makes it difficult to measure. However, copeptin (the C-terminal part of the pre-prohormone that is released with AVP) is a marker of AVP secretion because it is secreted along with AVP in equimolar amounts, and it is easier to monitor because its half-life is longer and it is a more stable protein [287].
Control of AVP synthesis and release
The gene encoding AVP is expressed in the large-diameter neurons of the supraoptic (SO) and paraventricular (PV) nuclei of the hypothalamus. This gene encodes a prohormone that must undergo specific proteolytic processing to produce the active AVP hormone. Thus, the AVP gene encodes three peptides: i) the 9–amino acid peptide arginine vasopressin, ii) a carrier protein called neurophysin-2 and iii) a small glycoprotein called copeptin.
Osmoreceptors are located in the organum vasculosum laminae terminalis and subfornical organ of the hypothalamus. These two areas breech the blood-brain barrier and are able to sense changes in plasma osmolality [288], and they have therefore been shown to be involved in controlling body water content. They respond to elevations in plasma osmolality by increasing the activity of mechanosensitive cation channels that are located in their cell membranes [289,290]. This results in significant membrane depolarization that increases the frequency of action potentials. These osmosensitive neurons project to the large-diameter neurons in the SO and PV nuclei of the posterior hypothalamus, and these neurons then synthesize AVP as a prohormone that is packaged in granules. By binding to the carrier protein neurohypophysin, these granules are transported along hypothalamic–neurohypophyseal tracts to the axon terminals of magnocellular neurons in the neurohypophysis, where the AVP is then stored. When stimulated by the osmosensitive neurons, these magnocellular neurons release stored AVP into the neurohypophysis – an area that also lacks a blood-brain barrier – and AVP enters the general circulation. Hypothalamic–neurohypophyseal tracts also innervate other areas of brain and the spinal cord, which enables AVP to exert its actions not only systemically but also locally in the brain and spinal cord.
Principal determinants of AVP plasma levels
The two major factors that control AVP release are osmotic and non-osmotic stimulation [291]. However, the osmotic and non-osmotic pathways independently enter the same AVP neurons in the SO and PV nuclei [292]. Nevertheless, the baro- and osmoreceptor pathways do not function in isolation because a reduction in plasma volume increases the sensitivity of osmoreceptors.
Osmotic AVP release: The most sensitive stimulus of the secretion of AVP is plasma osmolality. Under normal circumstances, the low concentration of AVP in the plasma is very sensitive, and it increases rapidly and linearly in response to very small changes in plasma osmolality [286,289]. However, this sensitivity is influenced by the nature of the solute, the rate of the change in plasma osmolality, and the age and alcohol intake of the individual [293]. A normal level of plasma osmolality is ~ 293 mOsm/kg, and when it increases by about 1% of this value, there is a rise in the: i) synthesis of AVP in the hypothalamus, ii) secretion of AVP from vasopressinergic nerve endings in the neurohypophysis, and iii) release of the AVP that was previously stored in the neurohypophysis into the general circulation. Recently, in a transgenic rat line expressing an AVP-eGFP fusion gene, the in vivo molecular processing of the AVP-eGFP fusion gene and the secretion of AVP that was stimulated by osmotic stimulation were demonstrated for the first time [294]. In addition, high plasma osmolality triggers thirst. However, the increased renal reabsorption of water free of solutes in response to AVP lowers plasma osmolality, reducing the stimulus for AVP secretion and thirst. The mechanisms that ensure water homeostasis and the fundamentals of water balance disorders were reviewed in detail in a recent publication [289].
Nonosmotic AVP release: the secretion of AVP is also influenced by changes in isotonic plasma volume (IPV) and blood pressure. Decreases in circulatory IPV and arterial blood pressure diminish the sensitivity of high-pressure and low-pressure (left atrial) baroreceptors and are potent nonosmotic stimuli for AVP release. Afferent fibers from arterial baroreceptors terminate in the nucleus of the tractus solitaries (NTS) of the dorsomedial medulla oblongata [295]. A1 adrenergic neurons in the ventrolateral medulla are involved in the afferent pathway from the NTS to the magnocellular neurons of the SO and PV nuclei in the hypothalamus, which synthesize AVP [296,297]. Furthermore, the baroreceptor-mediated afferent pathway for AVP release is activated by other factors, such as low cardiac output, left atrial distension, atrial tachycardia and hypoxia [291]. Nonosmotic AVP responses require larger variations than changes in plasma osmolality [298]. Therefore, reductions of 5% to 10% in IPV have little effect on AVP secretion, whereas reductions of 20% to 30% in IPV increase AVP plasma levels, causing them to reach 50 to 100 pg/ml [299]. At these high levels, AVP has potent V1a-mediated vasoconstrictor effects [300]. In the kidney, sympathetic activation induces i) arteriolar vasoconstriction, ii) a reduction in the glomerular filtration rate, iii) an increase in proximal Na+ and water reabsorption, with reduced distal delivery of water and Na+, and iv) an increase in distal Na+ and water reabsorption. In addition to baroreceptor activity, neurohumoral activation through ANG II increases the amount of AVP that is released in response to any given plasma osmolality [301]. Thus, a decrease in the effective circulatory blood volume impairs baroreceptor sensitivity, leading to an increase in the activity of the sympathetic nervous system, the activation of the RAS system and the nonosmotic release of AVP. Furthermore, AVP release is also stimulated by nausea and pain via central nervous input [302].
AVP acts via V1a, V1b, and V2 receptors that are located in the kidney and others regions.
V1 receptors: V1a receptors, which mediate the vasoconstrictor effect of AVP, are present in many tissues, including the smooth muscle cells of vessels and the brain, adrenal cortex, adipose tissue, and liver cells [285]. In the kidney, V1a receptors are localized in the renal vasculature, juxtaglomerular apparatus, macula densa cells, connecting tubule, collecting duct [303,304,305] and vasa recta [306]. AVP binding to the V1a receptor activates Gq/phospholipase C-b, resulting in an increase in [Ca2+]i levels and the activation of PKC, which causes vasoconstriction, platelet aggregation and the growth of smooth muscle cells [307,308]. V1b receptors are mainly present in the anterior pituitary, adrenal medulla, islet cells of Langerhans, and white adipose tissue [285]. Although the expression of the V1b receptor in the kidney has been described, its renal localization and functions are not well understood [309,310].
V2 receptors: As previously mentioned, the V2 receptor acts in the renal principal cells of the distal convoluted tubule, connecting tubule, collecting tubule and duct [305,311], and thick ascending limb of Henle [312]. It has been demonstrated that the human V2 receptor cDNA encodes 371 amino acids and a protein that has seven transmembrane domains, which is characteristic of G-protein-coupled receptors [314]. Thus, the V2 receptors have physiological functions that are mediated largely by the heterotrimeric G-protein Gs. These induce the activation of adenylyl cyclases to increase the intracellular level of cAMP [314] which activates PKA and is catabolized by cAMP-dependent phosphodiesterase. In turn, the phosphorylation of PKA mediates AVP cellular signaling to the aquaporin 2 water channel. This leads to i) the translocation of aquaporin 2 water channels from the membranes of cytoplasmic vesicles to the luminal tubular membrane, ii) an increase in the water permeability of these membranes, and iii) an increase in the expression of this water channel.
AVP Renal Actions on Na+reabsorption
AVP effects on Na+ reabsorption in the cortical collecting tubule: AVP up-regulates Na+ reabsorption in the cortical collecting tubule by stimulating ENaC channels [315,316]. This rapid AVP effect is the result of membrane trafficking of ENaC via the regulation of the ubiquitin ligase Nedd4-2 [317]. However, it is probable that AVP is also involved in the long-term regulation of ENaC in this renal segment [318].
AVP receptors involved in distal nephron HCO3- transport:
The cortical distal tubule of the mammalian kidney plays an important role in the renal control of HCO3- reabsorption and H+ secretion. Processes such as electroneutral Na+/H+ exchange (via the basolateral NHE1 isoform or the apical NHE3 isoform) and electrogenic H+ secretion (via H+-ATPase) in addition to H+/K+-ATPase and Cl−/ HCO3- exchange and Cl−channels play key roles in determining the rate and direction of bicarbonate transport in this nephron segment [319]. Although the major function of AVP in the kidney is to increase water permeability in the cortical collecting duct (CCD) and inner medullary collecting duct (IMCD) [320], AVP has also been shown to be involved in tubular fluid acidification. In the rat kidney, AVP inhibits bicarbonate reabsorption in the thick ascending limb [321,322] and stimulates proton secretion (or bicarbonate reabsorption) in the distal tubule [321] and CCD [321,323]. However, in A6 cells (an amphibian distal nephron cell line), adding AVP at either low (10-10 M) or high (10-6 M) concentrations inhibited basolateral NHE1 activity [192]. Studies in isolated perfused mouse medullary thick ascending limbs showed that AVP stimulated basolateral NHE1 while simultaneously inhibiting apical NHE3 [324]. Thus, it is possible that the difference in AVP responses in distal nephron HCO3- transport may vary according to the cell membrane type that is being studied. Since the concentration of AVP in luminal fluid may exceed 1,000-fold its concentration in plasma [325] and considering the potential existence of functional luminal AVP receptors [326,327], a study was conducted in vivo in early distal (ED) and late distal (LD) segments of the rat kidney to determine whether luminal AVP (10-9 M) regulates acid-base transport in the rat cortical distal tubule [115]. The results of this study indicated that AVP has a direct effect that stimulates net HCO3- reabsorption in the ED and LD segments by activating VI receptors at the luminal membrane because this stimulatory effect was prevented by simultaneous luminal perfusion with the V1 antagonist [(d (CH2)5, Tyr (Et)2) arginine vasopressin]. In addition, it was observed that luminal AVP exerts effects on [Ca2+]i in the medullary thick ascending limb [328] and CCD [329]. So, taken together, these findings suggest that luminal AVP might play a significant role in the regulation of HCO3- reabsorption in the distal convoluted, intercalated and initial collecting duct segments via V1 receptor-mediated mechanisms.
The possible physiological role of luminal AVP in distal nephron activities should also be considered. The concentration of AVP in the luminal fluid may be higher than that in plasma, even under physiological conditions. Data from kinetic studies and from studies of renal clearances have suggested that the tubular secretion of AVP into the distal nephron is an important component of renal clearance of this hormone in humans [325], dogs [330,331] and rats [332]. In humans, AVP clearance is in the range of 0.1 to 2 ml/min/kg, and urine AVP levels range from 5 to 500 p Mol [329]. In addition, it was reported [326] that water permeability stimulated by basolateral AVP was inhibited by luminal AVP within this concentration range and that luminal AVP at a concentration of 100 pMol increased [Ca2+]i [329]. These data collectively suggest that luminal AVP might play a significant role in regulating the tubule transport of water and ions, especially when the AVP concentration is high.
Most studies have analyzed AVP activity after it has been applied at the distal nephron basolateral surface. It’s activity is then mediated by V2 receptors via the adenylate cyclase/cAMP-protein kinase A signalling system, and this pathway, at high-AVP concentrations, is thought to inhibit the Na+/H+ exchanger [333]. However, V1 receptors have been detected in both apical and basolateral membrane domains, where they have been shown to mediate AVP activity via phospholipase C-IP3-calcium signalling [329,334]. However, it is known that PKC, when phosphorylated, may stimulate the Na+/H+ exchanger [92]. In addition, in MDCK cells, the stimulatory effect of AVP on NHE1 is mediated by the activation of V1 receptors that are located on the basolateral cell membrane surface, while basolateral V2 receptors have a dose-dependent inhibitory effect [335].
Thus, because the response to AVP involving distal bicarbonate reabsorption may vary with the hormonal dose being studied and the type of receptor that is present on the distal cell membrane surface, a study was performed [336] to determine whether perfusing AVP into peritubular capillaries at either low (10−11 M) or at high (10−9 M) concentrations would regulate bicarbonate transport in vivo in ED and LD segments in rats. In these experiments, the kinetics of HCO3- reabsorption were evaluated using an in vivo stopped-flow tubular microperfusion technique which it is not affected by the glomerular filtration rate [72]. During this research [336] the systemic alterations that are caused by microperfusing hormones into peritubular capillaries were avoided, as confirmed by an absence of changes in urine flow, Na+ excretion, and systemic acid-base alterations. Another advantage of using in vivo peritubular capillary AVP perfusion is that it allows the researcher to ensure that measurements of acidification kinetics are performed at well-determined hormonal levels because it has been shown that some peptides have a short half-life when injected parenterally. This is important because the intact structure of AVP is required for it to binding to its receptor [337]. To detect which specific basolateral receptor AVP regulates distal bicarbonate reabsorption, this study [336] examined the effects of the following V1 and V2 receptor-specific antagonists: 8-bromoadenosine 3′,5′-cyclic monophosphate (8-BrcAMP; a membrane-permeant cAMP analog) and deamino-Cys1,d-Arg8 vasopressin (dDAVP; a V2-selective agonist), respectively, and dDAVP (an AVP analog that specifically binds to adenylyl cyclase-coupled V2 receptors). In conclusion, this work [336] was the first to indicate that: 1) peritubular AVP (10−11 or 10−9 M) significantly stimulated HCO3- reabsorption in ED and LD segments by activating basolateral V1 receptors, 2) basolateral V2 receptors functions in a dose-dependent inhibitory effect that was mediated by cAMP, and 3) endogenous AVP stimulated bicarbonate reabsorption in ED and LD segments through a basolateral V1 receptor-mediated process.
AVP dose-dependent biphasic effect on Na+/H+ exchanger: Figure 5 demonstrates the effect of AVP (10-12, 10-9 or 10-6 M) on (pHi)r and [Ca2+]i in MDCK cells [335]. In control cells, the (pHi)r was 0.101 ± 0.005 pH units/min [79]. The addition of AVP (10-12 or 10-9 M) to the bath caused a significant increase in the (pHi)r (by 77% or 31%, respectively), but a lower concentration of AVP (10-6 M) resulted in a significant decrease (by 69%). ANP alone or dimethyl-BAPTA-AM alone did not affect the (pHi)r but impaired both the stimulatory and the inhibitory effect of AVP on it. Figure 5 also shows that MDCK cells exhibited a mean baseline [Ca2+]i of 99 ± 5 nM . The subsequent addition of AVP (10-12, 10-9 and 10-6 M) progressively increased [Ca2+]i from control values to 340 ± 4 nM (25) in a dose-dependent manner. The addition of ANP alone to the bathing solution led to a rapid and significant decrease in [Ca2+]i to 40 ± 4 nM [121], and the subsequent addition of AVP (10-12, 10-9 and 10-6 M) resulted in a recovery of [Ca2+]i that reached 80% without exceeding normal baseline values. The figure also shows that dimethyl-BAPTA-AM alone led to a significant decrease in [Ca2+]i (by 55%), and the subsequent addition of AVP (10-12, 10-9 and 10-6 M) resulted in a recovery of [Ca2+]i that did not achieve the respective control values. Hence, ANP and dimethyl-BAPTA-AM, which caused a moderate decrease in [Ca2+]i, did not affect the (pHi)r but impaired the pathway that caused an increase in [Ca2+]i, thereby blocking both the stimulatory and the inhibitory effect of AVP on this process. So, these studies are compatible with stimulation of the NHE1 exchanger by increases of [Ca2+]i in the lower range (that is, 10-12 or 10-9 M AVP; mediated by basolateral V1 receptors) and inhibition at high [Ca2+]i levels (10-6 M AVP; via activation of basolateral V1 receptors). They also are compatible with inhibition of the exchanger at high cell cAMP levels (at 10-6 M AVP, mediated by basolateral V2 receptors). Therefore, the biphasic action of AVP on the Na+/H+ exchanger is similar to the biphasic action described for ANG II (Figure 1) and ALDO (Figure 4).
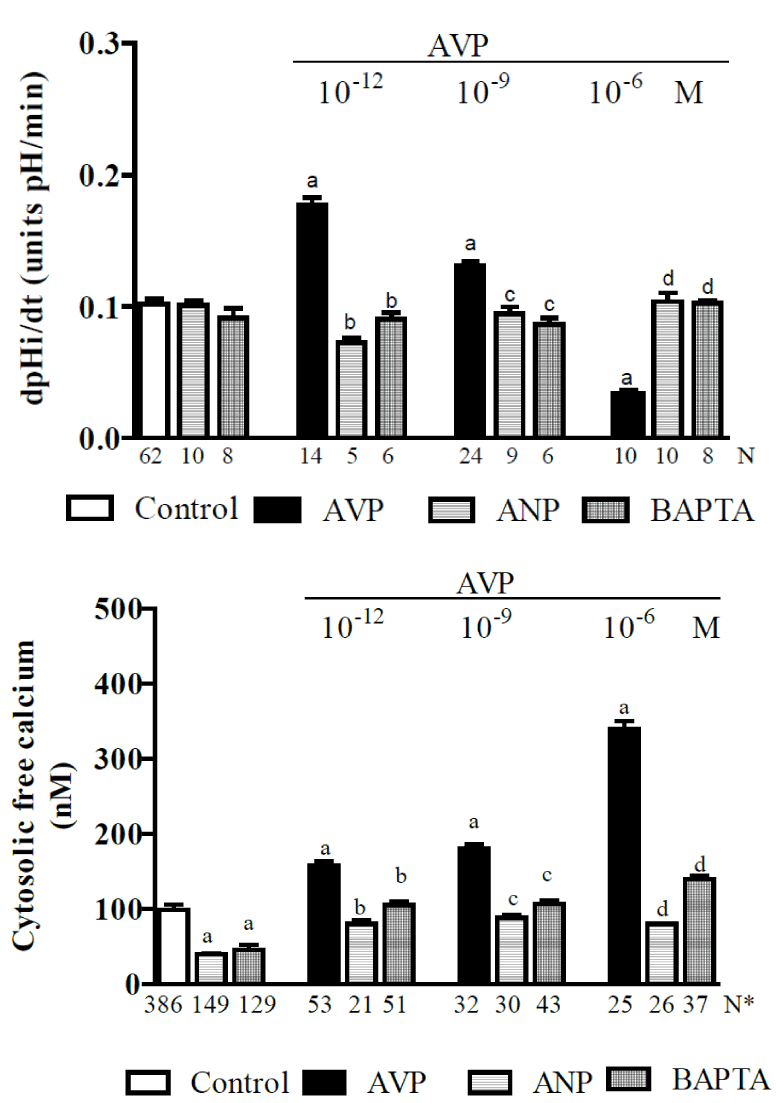
Figure 5. Research in MDCK cells [335] indicating the effects of AVP (10-12, 10-9 or 10-6 M) alone or plus ANP (10-6 M) or dimethyl-BAPTA/AM (5 X 10-5 M) on: A - pH intracellular recovery rates and B - cytosolic free calcium concentrations. Values are shown as the means ± SE. N = no. of observations. N* = no. of experiments in which the maximum fluorescent signal was averaged for 10 cells. dpHi/dt = pH intracellular recovery rate in the first 2 min after a NH4Cl pulse. a = P < 0.05 vs. control. b = P < 0.05 vs. AVP (10-12 M). c = P < 0.05 vs. AVP (10-9 M). d = P < 0.05 vs. AVP (10-6 M).
This dual regulation of the Na+/H+ exchanger (or bicarbonate reabsorption) by AVP may represent a relevant regulatory mechanism that prevents blood alkalinization under conditions involving volume depletion because it is known that plasma AVP levels are usually 20–30 times greater than normal when the blood volume is reduced by 20–30% (in rats, dogs, and humans) or the plasma osmolality is increased by 10% [338].
Figure 6 summarizes some of the data produced by our laboratory that have been discussed in this Review. These data have been combined in a schematic model that describes the biphasic effects of ANG II, ALDO, AVP and ANG-(1-7) on the calcium-dependent regulation of the Na+/H+ exchanger (NHE) by CaM. In the presence of basal activity, CaM binds at two sites to the cytosolic tail of the NHE: region A (a high affinity site that works as an autoinhibitory domain of the exchanger) and region B (a low affinity site that acts as an autostimulatory domain of the exchanger). A discrete increase in [Ca2+]i (caused by different hormonal concentrations, as indicated in Figure 6) may induce Ca2+/CaM to bin to region A, thus blocking the inhibitory interaction and activating the NHE. On the other hand, region B of the exchanger binds to Ca2+/CaM only when there has been a higher increase in [Ca2+]i (induced by various hormonal concentrations, as indicated in Figure 6), and under these conditions, binding inhibits the NHE. This scheme is based on theoretical studies [81,90] and on experimental studies using site-directed mutagenesis [18] it is known that the biphasic hormonal effects on the calcium-dependent regulation of NHE3 are dependent on calmodulin (CaM). Briefly, the data indicate that CaM binds to NHE1 at two sites in the cytosolic tail: region A (a high affinity site that contains residues 637-657) and region B (a low affinity site that is comprised of residues 657-700) [26]. Despite the fact that extensive research has been conducted to determine how binding between Ca2+/CaM and NHE1 occurs and the structure of the regulatory domain that is involved in its complex with CHP1 or CHP2 using nuclear magnetic resonance [32], X-ray crystallography [33] and, more recently, small angle X-ray scattering analysis [1], the binding mechanism by which Ca2+/CaM activates NHE1 is not easy to understand and is not yet satisfactorily or fully described. In section 2.1 NHE - NHE1 in this review, there is a presentation of the main proposals that are currently available. Additionally, under basal conditions, NHE3 is active, and CaMKII binds to the NHE3 C-terminal autoinhibitory domain (aa 586–605). This binding is rapidly reduced by a discrete increase in [Ca2+]i, which causes a conformational change in CaMKII that allows high-affinity interactions with target proteins and prevents the inactivation of the kinase by re-association between the catalytic domain and the autoinhibitory domain after Ca2+ returns to basal levels [13,47,48]. Other proteins that associate with NHE3 require this NHE3 domain, and these include NHERF1–4, phospholipase Cγ and CK2α [43,44]. The CaMKII-mediated inhibition of basal NHE3 activity is NHERF2-dependent, modifies NHE3 turnover rates, and causes the phosphorylation of NHE3. In addition, this regulatory effect requires amino acids in the C-terminal-binding autostimulatory domain that are downstream of NHE3 to interact with CaMKII (aa 690) [45,46]. Hence, CaMKII constitutively binds to, phosphorylates, and inhibits NHE3 via a NHERF2 protein-dependent process [48]. More details of these mechanisms are provided in section 2.1 NHE – NHE3 in the present Review. Therefore, the present results indicate stimulation of these exchangers by increases of [Ca2+]i in the lower range, and inhibition at high [Ca2+]i levels. In support of this idea, we have provided the following results: i) ANP and dimethyl-BAPTA-AM, by causing a moderate decrease in [Ca2+]i, do not affect the (pHi)r but impair the pathway that causes an increase in [Ca2+]i, thereby blocking both the stimulatory and the inhibitory effect of ANG II (Figure 1) or AVP (Figure 5) in this process; ii) EGTA alone causes a dramatic decline in [Ca2+]i, impairs the (pHi)r and blocks both the stimulatory and the inhibitory effect of ANG II on it (Figure 1); iii) Dimethyl-BAPTA-AM alone does not affect the JHCO3- but impairs both the inhibitory and the stimulatory effect of ANG-(1-7) on it (Figure 2); and iv) thapsigargin alone causes a dramatic increase in [Ca2+]I, inhibits the JHCO3- and impairs both the inhibitory and the stimulatory effect of ANG-(1-7) on it (Figure 2). Determining whether modifications of the [Ca2+]i represent an important direct mechanism for altering exchanger activity or a side-effect of other signalling pathways will require additional studies.
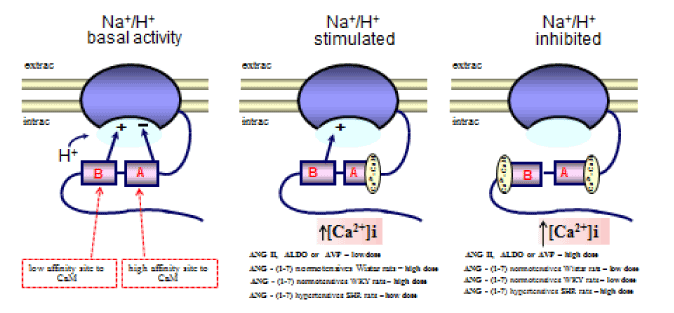
Figure 6. Schematic model indicating the biphasic effects of ANG II, ANG(1-7), ALDO and AVP on the calcium-dependent regulation of the Na+/H+ exchanger by Calmodulin. CaM = Calmodulin. [Ca2+]i = cytosolic free calcium concentration. ↑ [Ca2+]i = discrete increase in [Ca2+]i. ↑[Ca2+]i = high increase in [Ca2+]i. ▬ = inhibition. + = stimulation. Extrac = extracellular. Intrac = intracellular. The details of the mechanisms involved are discussed in sections 2.1 NHE and 3. Summary in this Review.
In conclusion, taken together, the data described in this review are compatible with the hormonal stimulation of NHE1 or NHE3 being induced by increases in [Ca2+]i at lower concentrations and inhibited at high [Ca2+]i levels. In MDCK cells or in the proximal tubule in Wistar rats, ANG II, ALDO (via genomic and nongenomic effects) or AVP stimulate these exchangers at low doses and inhibit them at high doses. However, ANG–(1-7), in Wistar or WKY (normotensives) rats, has inverse dose-dependent effects. Moreover, in SHR (hypertensive) rats, the biphasic effect of ANG-(1-7) is similar to that of ANG II, ALDO or AVP in Wistar rats. The interactions between these hormonal effects may represent a regulatory mechanism of extracellular volume in normotensives animals. Accordingly, in hypertensives animals, a high plasma level of ANG-(1-7) inhibits NHE3 in the proximal tubule, which mitigates hypertension.
- Köster S, Pavkov-Keller T, Kühlbrandt W, Yildiz Ö (2011) Structure of human Na+/H+ exchanger NHE1 regulatory region in complex with calmodulin and Ca2+. J Biol Chem 286: 40954-40961. [Crossref]
- Lee SH, Kim T, Park ES, Yang S, Jeong D, et al. (2008) NHE10, an osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival [corrected]. Biochem Biophys Res Commun 369: 320-326. [Crossref]
- Orlowski J, Grinstein S (2004) Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch 447: 549-565. [Crossref]
- Bobulescu IA, Moe OW (2009) Luminal Na(+)/H (+) exchange in the proximal tubule. Pflugers Arch 458: 5-21. [Crossref]
- Donowitz M, Li X (2007) Regulatory binding partners and complexes of NHE3. Physiol Rev 87: 825-872. [Crossref]
- Lee BL, Sykes BD, Fliegel L (2013) Structural and functional insights into the cardiac Naâº/H⺠exchanger. J Mol Cell Cardiol 61: 60-67. [Crossref]
- Li X, Prins D, Michalak M, Fliegel L (2013) Calmodulin-dependent binding to the NHE1 cytosolic tail mediates activation of the Na+/H+ exchanger by Ca2+ and endothelin. Am J Physiol Cell Physiol 305: C1161-1169. [Crossref]
- Shimada-Shimizu N, Hisamitsu T, Nakamura TY, Hirayama N, Wakabayashi S (2014) Na+/H+ exchanger 1 is regulated via its lipid-interacting domain, which functions as a molecular switch: a pharmacological approach using indolocarbazole compounds. Mol Pharmacol 85: 18-28. [Crossref]
- Singh V, Lin R, Yang J, Cha B, Sarker R, et al. (2014) AKT and GSK-3 are necessary for direct ezrin binding to NHE3 as part of a C-terminal stimulatory complex: role of a novel Ser-rich NHE3 C-terminal motif in NHE3 activity and trafficking. J Biol Chem 289:5449-5461. [Crossref]
- Slepkov ER, Rainey JK, Sykes BD, Fliegel L (2007) Structural and functional analysis of the Na+/H+ exchanger. Biochem J 401: 623-633. [Crossref]
- Wakabayashi S, Bertrand B, Ikeda T, Pouysségur J, Shigekawa M (1994) Mutation of calmodulin-binding site renders the Na+/H+ exchanger (NHE1) highly H(+)-sensitive and Ca2+ regulation-defective. J Biol Chem 269: 13710-13715. [Crossref]
- Wakabayashi S, Hisamitsu T, Nakamura TY (2013) Regulation of the cardiac Naâº/H⺠exchanger in health and disease. J Mol Cell Cardiol 61: 68-76. [Crossref]
- Zachos NC, Tse M, Donowitz M (2005) Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 67: 411-443. [Crossref]
- Grinstein S, Rotin D, Mason MJ (1989) Na+/H+ exchange and growth factor-induced cytosolic pH changes. Role in cellular proliferation. Biochim Biophys Acta 988: 73-97. [Crossref]
- Pouysségur J, Franchi A, L'Allemain G, Paris S (1985) Cytoplasmic pH, a key determinant of growth factor-induced DNA synthesis in quiescent fibroblasts. FEBS Lett 190: 115-119. [Crossref]
- Sardet C, Franchi A, Pouysségur J (1989) Molecular cloning, primary structure, and expression of the human growth factor-activatable Na+/H+ antiporter. Cell 56: 271-280. [Crossref]
- Goswami P, Paulino C, Hizlan D, Vonck J, Yildiz O, et al. (2011) Structure of the archaeal Na+/H+ antiporter NhaP1 and functional role of transmembrane helix 1. EMBO J 30: 439-449. [Crossref]
- Wakabayashi S, Fafournoux P, Sardet C, Pouysségur J (1992) The Na+/H+ antiporter cytoplasmic domain mediates growth factor signals and controls "H(+)-sensing". Proc Natl Acad Sci U S A 89: 2424-2428. [Crossref]
- Malo ME, Li L, and Fliegel L. (2007) Mitogen-activated protein kinase-dependent activation of the Na+/H+ exchanger is mediated through phosphorylation of amino acids Ser770 and Ser771. J Biol Chem 6292-6299. [Crossref]
- Khaled AR, Moor AN, Li A, Kim K, Ferris DK, et al. (2001) Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol 21: 7545-7557. [Crossref]
- Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, et al. (1999) p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem 274: 20206-20214. [Crossref]
- Baumgartner M, Patel H, Barber DL (2004) Na(+)/H(+) exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am J Physiol Cell Physiol 287: C844-850. [Crossref]
- Lin X, Barber DL (1996) A calcineurin homologous protein inhibits GTPase-stimulated Na-H exchange. Proc Natl Acad Sci U S A 93: 12631-12636. [Crossref]
- Pang T, Wakabayashi S, Shigekawa M (2002) Expression of calcineurin B homologous protein 2 protects serum deprivation-induced cell death by serum-independent activation of Na+/H+ exchanger. J Biol Chem 277: 43771-43777. [Crossref]
- Zaun HC, Shrier A, and Orlowski J. (2008) Calcineurin B homologous protein 3 promotes the biosynthetic maturation, cell surface stability, and optimal transport of the Na+/H+ exchanger NHE1 isoform. J Biol Chem 283: 12456-12467. [Crossref]
- Bertrand B, Wakabayashi S, Ikeda T, Pouyssegur J, and Shigekawa M. (1994) The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins. Identification and characterization of calmodulin-binding sites. J Biol Chem 269: 13703-13709. [Crossref]
- Aronson PS, Nee J, Suhm MA (1982) Modifier role of internal H+ in activating the Na+-H+ exchanger in renal microvillus membrane vesicles. Nature 299: 161-163. [Crossref]
- Lacroix J, Poët M, Maehrel C, Counillon L (2004) A mechanism for the activation of the Na/H exchanger NHE-1 by cytoplasmic acidification and mitogens. EMBO Rep 5: 91-96. [Crossref]
- O'Neil KT, DeGrado WF (1990) How calmodulin binds its targets: sequence independent recognition of amphiphilic alpha-helices. Trends Biochem Sci 15: 59-64. [Crossref]
- Sanyal G, Richard LM, Carraway KL 3rd, Puett D (1988) Binding of amphiphilic peptides to a carboxy-terminal tryptic fragment of calmodulin. Biochemistry 27: 6229-6236. [Crossref]
- Wakabayashi S, Ikeda T, Iwamoto T, Pouysségur J, Shigekawa M (1997) Calmodulin-binding autoinhibitory domain controls "pH-sensing" in the Na+/H+ exchanger NHE1 through sequence-specific interaction. Biochemistry 36: 12854-12861. [Crossref]
- Mishima M, Wakabayashi S, Kojima C (2007) Solution structure of the cytoplasmic region of Na+/H+ exchanger 1 complexed with essential cofactor calcineurin B homologous protein 1. J Biol Chem 282: 2741-2751. [Crossref]
- Ammar YB, Takeda S, Hisamitsu T, Mori H, Wakabayashi S (2006) Crystal structure of CHP2 complexed with NHE1-cytosolic region and an implication for pH regulation. EMBO J 25: 2315-2325. [Crossref]
- Orlowski J, Kandasamy RA, Shull GE (1992) Molecular cloning of putative members of the Na/H exchanger gene family. cDNA cloning, deduced amino acid sequence, and mRNA tissue expression of the rat Na/H exchanger NHE-1 and two structurally related proteins. J Biol Chem 267: 9331-9339. [Crossref]
- Tse CM, Brant SR, Walker MS, Pouyssegur J, Donowitz M (1992) Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney-specific Na+/H+ exchanger isoform (NHE-3). J Biol Chem 267: 9340-9346. [Crossref]
- Amemiya M, Loffing J, Lötscher M, Kaissling B, Alpern RJ, et al. (1995) Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney Int 48: 1206-1215. [Crossref]
- Biemesderfer D, Pizzonia J, Abu-Alfa A, Exner M, Reilly R, et al. (1993) NHE3: a Na+/H+ exchanger isoform of renal brush border. Am J Physiol 265: F736-742. [Crossref]
- Biemesderfer D, Rutherford PA, Nagy T, Pizzonia JH, Abu-Alfa AK, et al. (1997) Monoclonal antibodies for high-resolution localization of NHE3 in adult and neonatal rat kidney. Am J Physiol 273: F289-299. [Crossref]
- Lamprecht G, Heil A, Baisch S, Lin-Wu E, Yun CC, et al. (2002) The down regulated in adenoma (dra) gene product binds to the second PDZ domain of the NHE3 kinase A regulatory protein (E3KARP), potentially linking intestinal Cl-/HCO3- exchange to Na+/H+ exchange. Biochemistry 41: 12336-12342. [Crossref]
- Melvin JE, Park K, Richardson L, Schultheis PJ, Shull GE (1999) Mouse down-regulated in adenoma (DRA) is an intestinal Cl(-)/HCO(3)(-) exchanger and is up-regulated in colon of mice lacking the NHE3 Na(+)/H(+) exchanger. J Biol Chem 274: 22855-22861. [Crossref]
- Ahn W, Kim KH, Lee JA, Kim JY, Choi JY, et al. (2001) Regulatory interaction between the cystic fibrosis transmembrane conductance regulator and HCO3- salvage mechanisms in model systems and the mouse pancreatic duct. J Biol Chem 276: 17236-17243. [Crossref]
- Thwaites DT, Kennedy DJ, Raldua D, Anderson CM, Mendoza ME, et al. (2002) H/dipeptide absorption across the human intestinal epithelium is controlled indirectly via a functional Na/H exchanger. Gastroenterology 122: 1322-1333. [Crossref]
- Sarker R, Grønborg M, Cha B, Mohan S, Chen Y, et al. (2008) Casein kinase 2 binds to the C terminus of Na+/H+ exchanger 3 (NHE3) and stimulates NHE3 basal activity by phosphorylating a separate site in NHE3. Mol Biol Cell 19: 3859-3870. [Crossref]
- Zachos NC, van Rossum DB, Li X, Caraveo G, Sarker R, et al. (2009) Phospholipase C-gamma binds directly to the Na+/H+ exchanger 3 and is required for calcium regulation of exchange activity. J Biol Chem 19437-19444. [Crossref]
- Yang J, Singh V, Cha B, Chen TE, Sarker R, et al. (2013) NHERF2 protein mobility rate is determined by a unique C-terminal domain that is also necessary for its regulation of NHE3 protein in OK cells. J Biol Chem 288: 16960-16974. [Crossref]
- Zachos NC, Kovbasnjuk O, Donowitz M (2009) Regulation of intestinal electroneutral sodium absorption and the brush border Na+/H+ exchanger by intracellular calcium. Ann N Y Acad Sci 1165: 240-248. [Crossref]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, et al. (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133: 462-474. [Crossref]
- Zizak M, Chen T, Bartonicek D, Sarker R, Zachos NC, et al. (2012) Calmodulin kinase II constitutively binds, phosphorylates, and inhibits brush border Na+/H+ exchanger 3 (NHE3) by a NHERF2 protein-dependent process. J Biol Chem 13442-13456. [Crossref]
- Griffith LC. (2004) Regulation of calcium/calmodulin-dependent protein kinase II activation by intramolecular and intermolecular interactions. J Neurosci 24: 8394-8398. [Crossref]
- Yang E, and Schulman H. (1999) Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J Biol Chem 274: 26199-26208. [Crossref]
- Bobulescu IA, Di Sole F, Moe OW (2005) Na+/H+ exchangers: physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens 14: 485-494. [Crossref]
- Castrop H, Höcherl K, Kurtz A, Schweda F, Todorov V, et al. (2010) Physiology of kidney renin. Physiol Rev 90: 607-673. [Crossref]
- MacKenzie SM, Clark CJ, Fraser R, Gómez-Sánchez CE, Connell JM, et al. (2000) Expression of 11beta-hydroxylase and aldosterone synthase genes in the rat brain. J Mol Endocrinol 24: 321-328. [Crossref]
- Paul M, Wagner J, and Dzau VJ. (1993) Gene expression of the renin-angiotensin system in human tissues. Quantitative analysis by the polymerase chain reaction. J Clin Invest 91: 2058-2064. [Crossref]
- Prabhakar SS (2004) Regulatory and functional interaction of vasoactive factors in the kidney and extracellular pH. Kidney Int 66: 1742-1754. [Crossref]
- Dzau VJ (2001) Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 37: 1047-1052. [Crossref]
- Inagami T, Guo DF, Kitami Y (1994) Molecular biology of angiotensin II receptors: an overview. J Hypertens Suppl 12: S83-94. [Crossref]
- Mehta PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82-97. [Crossref]
- Ichiki T, Labosky PA, Shiota C, Okuyama S, Imagawa Y, et al. (1995) Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature 377: 748-750. [Crossref]
- Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, et al. (2006) Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A 103: 17985-17990. [Crossref]
- Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, et al. (2005) Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 115: 1092-1099. [Crossref]
- Schmitz U, and Berk BC. Angiotensin II signal transduction: Stimulation of multiple mitogen-activated protein kinase pathways. Trends Endocrinol Metab 8: 261-266, 1997.
- Ushio-Fukai M, Alexander RW, Akers M, Lyons PR, Lassègue B, et al. (1999) Angiotensin II receptor coupling to phospholipase D is mediated by the betagamma subunits of heterotrimeric G proteins in vascular smooth muscle cells. Mol Pharmacol 55: 142-149. [Crossref]
- Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, et al. (1993) Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem 268: 24539-24542. [Crossref]
- Shanmugam S, Corvol P, Gasc JM (1996) Angiotensin II type 2 receptor mRNA expression in the developing cardiopulmonary system of the rat. Hypertension 28: 91-97. [Crossref]
- Shanmugam S, Sandberg K (1996) Ontogeny of angiotensin II receptors. Cell Biol Int 20: 169-176. [Crossref]
- Steckelings UM, Kaschina E, Unger T (2005) The AT2 receptor--a matter of love and hate. Peptides 26: 1401-1409. [Crossref]
- Widdop RE, Jones ES, Hannan RE, Gaspari TA (2003) Angiotensin AT2 receptors: cardiovascular hope or hype? Br J Pharmacol 140: 809-824. [Crossref]
- Boron WF (2006) Acid-base transport by the renal proximal tubule. J Am Soc Nephrol 17: 2368-2382. [Crossref]
- Alpern RJ (1990) Cell mechanisms of proximal tubule acidification. Physiol Rev 70: 79-114. [Crossref]
- Liu FY, Cogan MG (1989) Angiotensin II stimulates early proximal bicarbonate absorption in the rat by decreasing cyclic adenosine monophosphate. J Clin Invest 84: 83-91. [Crossref]
- Malnic G, de Mello-Aires M (1971) Kinetic study of bicarbonate reabsorption in proximal tubule of the rat. Am J Physiol 220: 1759-1767. [Crossref]
- Gomes GN, Aires MM (1992) Interaction of atrial natriuretic factor and angiotensin II in proximal HCO3- reabsorption. Am J Physiol 262: F303-308. [Crossref]
- Schelling JR, Singh H, Marzec R, Linas SL (1994) Angiotensin II-dependent proximal tubule sodium transport is mediated by cAMP modulation of phospholipase C. Am J Physiol 267: C1239-1245. [Crossref]
- Cano A, Miller RT, Alpern RJ, Preisig PA (1994) Angiotensin II stimulation of Na-H antiporter activity is cAMP independent in OKP cells. Am J Physiol 266: C1603-1608. [Crossref]
- Du Z, Ferguson W, Wang T (2003) Role of PKC and calcium in modulation of effects of angiotensin II on sodium transport in proximal tubule. Am J Physiol Renal Physiol 284: F688-692. [Crossref]
- Liu FY, Cogan MG (1990) Role of protein kinase C in proximal bicarbonate absorption and angiotensin signaling. Am J Physiol 258: F927-933. [Crossref]
- Jourdain M, Amiel C, Friedlander G (1992) Modulation of Na-H exchange activity by angiotensin II in opossum kidney cells. Am J Physiol 263: C1141-1146. [Crossref]
- Coppola S, and Frömter E. (1994) An electrophysiological study of angiotensin II regulation of Na-HCO3 cotransport and K conductance in renal proximal tubules. I. Effect of picomolar concentrations. Pflugers Arch 427: 143-150. [Crossref]
- Geibel J, Giebisch G, Boron WF (1990) Angiotensin II stimulates both Na(+)-H+ exchange and Na+/HCO3- cotransport in the rabbit proximal tubule. Proc Natl Acad Sci U S A 87: 7917-7920. [Crossref]
- Ruiz OS, Qiu YY, Wang LJ, Arruda JA (1995) Regulation of the renal Na-HCO3 cotransporter: IV. Mechanisms of the stimulatory effect of angiotensin II. J Am Soc Nephrol 6: 1202-1208. [Crossref]
- Harris PJ (1992) Regulation of proximal tubule function by angiotensin. Clin Exp Pharmacol Physiol 19: 213-222. [Crossref]
- Houillier P, Chambrey R, Achard JM, Froissart M, Poggioli J, et al. (1996) Signaling pathways in the biphasic effect of angiotensin II on apical Na/H antiport activity in proximal tubule. Kidney Int 50: 1496-1505. [Crossref]
- Coppola S, and Frömter E. (1994) An electrophysiological study of angiotensin II regulation of Na-HCO3 cotransport and K conductance in renal proximal tubules. II. Effect of micromolar concentrations. Pflugers Arch 427: 151-156. [Crossref]
- Banday AA, and Lokhandwala MF. (2008) Loss of biphasic effect on Na/K-ATPase activity by angiotensin II involves defective angiotensin type 1 receptor-nitric oxide signaling. Hypertension 52: 1099-1105. [Crossref]
- Navar LG, Harrison-Bernard LM, Wang CT, Cervenka L, Mitchell KD (1999) Concentrations and actions of intraluminal angiotensin II. J Am Soc Nephrol 10 Suppl 11: S189-195. [Crossref]
- Haithcock D, Jiao H, Cui XL, Hopfer U, Douglas JG (1999) Renal proximal tubular AT2 receptor: signaling and transport. J Am Soc Nephrol 10 Suppl 11: S69-74. [Crossref]
- Poggioli J, Lazar G, Houillier P, Gardin JP, Achard JM, et al. (1992) Effects of angiotensin II and nonpeptide receptor antagonists on transduction pathways in rat proximal tubule. Am J Physiol 263: C750-758. [Crossref]
- Horita S, Zheng Y, Hara C, Yamada H, Kunimi M, et al. (2002) Biphasic regulation of Na+-HCO3- cotransporter by angiotensin II type 1A receptor. Hypertension 40: 707-712. [Crossref]
- Li Y, Yamada H, Kita Y, Kunimi M, Horita S, et al. (2008) Roles of ERK and cPLA2 in the angiotensin II-mediated biphasic regulation of Na+-HCO3(-) transport. J Am Soc Nephrol 19: 252-259. [Crossref]
- Zheng Y, Horita S, Hara C, Kunimi M, Yamada H, et al. (2003) Biphasic regulation of renal proximal bicarbonate absorption by luminal AT(1A) receptor. J Am Soc Nephrol 14: 1116-1122. [Crossref]
- Douglas JG, Hopfer U (1994) Novel aspect of angiotensin receptors and signal transduction in the kidney. Annu Rev Physiol 56: 649-669. [Crossref]
- Gunasegaram S, Haworth RS, Hearse DJ, Avkiran M (1999) Regulation of sarcolemmal Na(+)/H(+) exchanger activity by angiotensin II in adult rat ventricular myocytes: opposing actions via AT(1) versus AT(2) receptors. Circ Res 85: 919-930. [Crossref]
- Van Obberghen-Schilling E, Chambard JC, Paris S, L'Allemain G, and Pouysségur J. (1985) alpha-Thrombin-induced early mitogenic signalling events and G0 to S-phase transition of fibroblasts require continual external stimulation. EMBO J 4: 2927-2932. [Crossref]
- Carraro-Lacroix LR, Malnic G, Girardi AC (2009) Regulation of Na+/H+ exchanger NHE3 by glucagon-like peptide 1 receptor agonist exendin-4 in renal proximal tubule cells. Am J Physiol Renal Physiol 297: F1647-1655. [Crossref]
- Orlowski J, Kandasamy RA (1996) Delineation of transmembrane domains of the Na+/H+ exchanger that confer sensitivity to pharmacological antagonists. J Biol Chem 271: 19922-19927. [Crossref]
- Saccomani G, Mitchell KD, Navar LG (1990) Angiotensin II stimulation of Na(+)-H+ exchange in proximal tubule cells. Am J Physiol 258: F1188-1195. [Crossref]
- Woodcock EA, Johnston CI (1982) Inhibition of adenylate cyclase by angiotensin II in rat renal cortex. Endocrinology 111: 1687-1691. [Crossref]
- Douglas JG (1987) Angiotensin receptor subtypes of the kidney cortex. Am J Physiol 253: F1-7. [Crossref]
- He P, Klein J, and Yun CC. (2010) Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 27869-27878. [Crossref]
- Kwon TH, Nielsen J, Kim YH, Knepper MA, Frøkiaer J, et al. (2003) Regulation of sodium transporters in the thick ascending limb of rat kidney: response to angiotensin II. Am J Physiol Renal Physiol 285: F152-165. [Crossref]
- Nagami GT (2002) Enhanced ammonia secretion by proximal tubules from mice receiving NH(4)Cl: role of angiotensin II. Am J Physiol Renal Physiol 282: F472-477. [Crossref]
- Wall SM, Fischer MP, Glapion DM, De La Calzada M (2003) ANG II reduces net acid secretion in rat outer medullary collecting duct. Am J Physiol Renal Physiol 285: F930-937. [Crossref]
- Wang T, Giebisch G (1996) Effects of angiotensin II on electrolyte transport in the early and late distal tubule in rat kidney. Am J Physiol 271: F143-149. [Crossref]
- Weiner ID, New AR, Milton AE, Tisher CC (1995) Regulation of luminal alkalinization and acidification in the cortical collecting duct by angiotensin II. Am J Physiol 269: F730-738. [Crossref]
- Shirai A, Yamazaki O, Horita S, Nakamura M, Satoh N, et al. (2014) Angiotensin II dose-dependently stimulates human renal proximal tubule transport by the nitric oxide/guanosine 3',5'-cyclic monophosphate pathway. J Am Soc Nephrol 25: 1523-1532. [Crossref]
- Nakamura M, Shirai A, Yamazaki O, Satoh N, Suzuki M, et al. (2014) Roles of renal proximal tubule transport in acid/base balance and blood pressure regulation. Biomed Res Int 2014: 504808. [Crossref]
- Good DW, George T, Wang DH (1999) Angiotensin II inhibits HCO-3 absorption via a cytochrome P-450-dependent pathway in MTAL. Am J Physiol 276: F726-736. [Crossref]
- Barreto-Chaves ML, Mello-Aires M (1996) Effect of luminal angiotensin II and ANP on early and late cortical distal tubule HCO3- reabsorption. Am J Physiol 271: F977-984. [Crossref]
- Wagner CA, Mohebbi N, Uhlig U, Giebisch GH, Breton S, et al. (2011) Angiotensin II stimulates H?-ATPase activity in intercalated cells from isolated mouse connecting tubules and cortical collecting ducts. Cell Physiol Biochem 28: 513-520. [Crossref]
- Pech V, Zheng W, Pham TD, Verlander JW, Wall SM (2008) Angiotensin II activates H+-ATPase in type A intercalated cells. J Am Soc Nephrol 19: 84-91. [Crossref]
- Sun P, Yue P, Wang WH (2012) Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol 302: F679-687. [Crossref]
- Oliveira-Souza M, De Mello-Aires M (2000) Interaction of angiotensin II and atrial natriuretic peptide on pH(i) regulation in MDCK cells. Am J Physiol Renal Physiol 279: F944-953. [Crossref]
- Arima S (2003) Role of angiotensin II and endogenous vasodilators in the control of glomerular hemodynamics. Clin Exp Nephrol 7: 172-178. [Crossref]
- Barreto-Chaves ML, and de Mello-Aires M. (1997) Luminal arginine vasopressin stimulates Na(+)-H+ exchange and H(+)-ATPase in cortical distal tubule via V1 receptor. Kidney Int 52: 1035-1041. [Crossref]
- Musa-Aziz R, and Mello-Aires M. (2005) Action of ANG II and ANP on colon epithelial cells. Pflugers Arch 450: 405-414. [Crossref]
- Eguti DM, Thieme K, Leung GP, Mello-Aires M, and Oliveira-Souza M. (2010) Regulation of Na+/H+ exchanger isoform 1 (NHE1) by calmodulin-binding sites: role of angiotensin II. Cell Physiol Biochem. 541-552. [Crossref]
- Costa-Pessoa JM, Figueiredo CF, Thieme K, and Oliveira-Souza M. (2013) The regulation of NHE1 and NHE3 activity by angiotensin II is mediated by the activation of the angiotensin II type I receptor/phospholipase C/calcium/calmodulin pathway in distal nephron cells. Eur J Pharmacol 721: 322-331. [Crossref]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, et al. (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87: E1-9. [Crossref]
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, et al. (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275: 33238-33243. [Crossref]
- Jankowski V, Vanholder R, van der Giet M, Tölle M, Karadogan S, et al. (2007) Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol 27: 297-302. [Crossref]
- Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, et al. (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417-1427. [Crossref]
- Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, et al. (2003) Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A 100: 8258-8263. [Crossref]
- Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, et al. (2013) Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res 112: 1104-1111. [Crossref]
- Haulica I, Bild W, Serban DN (2005) Angiotensin peptides and their pleiotropic actions. J Renin Angiotensin Aldosterone Syst 6: 121-131. [Crossref]
- Pendergrass KD, Averill DB, Ferrario CM, Diz DI, Chappell MC (2006) Differential expression of nuclear AT1 receptors and angiotensin II within the kidney of the male congenic mRen2. Lewis rat. Am J Physiol Renal Physiol 290: F1497-1506. [Crossref]
- Danilczyk U, Pennin2021 Copyright OAT. All rights reservenzyme II in the heart and the kidney. Circ Res 98: 463-471. [Crossref]
- Ferrario CM, Trask AJ, Jessup JA (2005) Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol 289: H2281-2290. [Crossref]
- Keidar S, Strizevsky A, Raz A, Gamliel-Lazarovich A (2007) ACE2 activity is increased in monocyte-derived macrophages from prehypertensive subjects. Nephrol Dial Transplant 22: 597-601. [Crossref]
- Lambert DW, Hooper NM, Turner AJ (2008) Angiotensin-converting enzyme 2 and new insights into the renin-angiotensin system. Biochem Pharmacol 75: 781-786. [Crossref]
- Santos RA (2014) Angiotensin-(1-7). Hypertension 63: 1138-1147. [Crossref]
- Santos RA, Ferreira AJ, Simões E Silva AC (2008) Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol 93: 519-527. [Crossref]
- Varagic J, Trask AJ, Jessup JA, Chappell MC, Ferrario CM (2008) New angiotensins. J Mol Med (Berl) 86: 663-671. [Crossref]
- Jackson TR, Blair LA, Marshall J, Goedert M, Hanley MR (1988) The mas oncogene encodes an angiotensin receptor. Nature 335: 437-440. [Crossref]
- Metzger R, Bader M, Ludwig T, Berberich C, Bunnemann B, et al. (1995) Expression of the mouse and rat mas proto-oncogene in the brain and peripheral tissues. FEBS Lett 357: 27-32. [Crossref]
- Young D, O'Neill K, Jessell T, Wigler M (1988) Characterization of the rat mas oncogene and its high-level expression in the hippocampus and cerebral cortex of rat brain. Proc Natl Acad Sci U S A 85: 5339-5342. [Crossref]
- Becker LK, Etelvino GM, Walther T, Santos RA, and Campagnole-Santos MJ. (2007) Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am J Physiol Heart Circ Physiol 293: H1416-1424. [Crossref]
- Peiró C, Vallejo S, Gembardt F, Azcutia V, Heringer-Walther S, et al. (2007) Endothelial dysfunction through genetic deletion or inhibition of the G protein-coupled receptor Mas: a new target to improve endothelial function. J Hypertens 25: 2421-2425. [Crossref]
- Xu P, Costa-Goncalves AC, Todiras M, Rabelo LA, Sampaio WO, et al. (2008) Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension 51: 574-580. [Crossref]
- Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, et al. (2006) Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension 47: 996-1002. [Crossref]
- Giani JF, Gironacci MM, Muñoz MC, Peña C, Turyn D, et al. (2007) Angiotensin-(1 7) stimulates the phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: role of the AT1 and Mas receptors. Am J Physiol Heart Circ Physiol 293: H1154-1163. [Crossref]
- Dias-Peixoto MF, Santos RA, Gomes ER, Alves MN, Almeida PW, et al. (2008) Molecular mechanisms involved in the angiotensin-(1-7)/Mas signaling pathway in cardiomyocytes. Hypertension 52: 542-548. [Crossref]
- Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, et al. (2007) Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 49: 185-192. [Crossref]
- Su Z, Zimpelmann J, Burns KD (2006) Angiotensin-(1-7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int 69: 2212-2218. [Crossref]
- Tallant EA, Ferrario CM, Gallagher PE (2005) Angiotensin-(1-7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol 289: H1560-1566. [Crossref]
- Mahon JM, Carr RD, Nicol AK, Henderson IW (1994) Angiotensin(1-7) is an antagonist at the type 1 angiotensin II receptor. J Hypertens 12: 1377-1381. [Crossref]
- Rowe BP, Saylor DL, Speth RC, Absher DR (1995) Angiotensin-(1-7) binding at angiotensin II receptors in the rat brain. Regul Pept 56: 139-146. [Crossref]
- Walters PE, Gaspari TA, Widdop RE (2005) Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 45: 960-966. [Crossref]
- Flores-Muñoz M, Smith NJ, Haggerty C, Milligan G, Nicklin SA (2011) Angiotensin1-9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J Physiol 589: 939-951. [Crossref]
- Gembardt F, van Veghel R, Coffman TM, Schultheiss HP, Danser AH, et al. (2012) Hemodynamic effects of vasorelaxant compounds in mice lacking one, two or all three angiotensin II receptors. Hypertens Res 35: 547-551. [Crossref]
- Sumners C, Horiuchi M, Widdop RE, McCarthy C, Unger T, et al. (2013) Protective arms of the renin-angiotensin-system in neurological disease. Clin Exp Pharmacol Physiol 40: 580-588.
- Durand MJ, Raffai G, Weinberg BD, Lombard JH (2010) Angiotensin-(1-7) and low-dose angiotensin II infusion reverse salt-induced endothelial dysfunction via different mechanisms in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol 299: H1024-1033. [Crossref]
- Pinheiro SV, Simões e Silva AC, Sampaio WO, de Paula RD, Mendes EP, et al. (2004) Nonpeptide AVE 0991 is an angiotensin-(1-7) receptor Mas agonist in the mouse kidney. Hypertension 44: 490-496. [Crossref]
- Raffai G, Durand MJ, Lombard JH (2011) Acute and chronic angiotensin-(1-7) restores vasodilation and reduces oxidative stress in mesenteric arteries of salt-fed rats. Am J Physiol Heart Circ Physiol 301: H1341-1352. [Crossref]
- Steckelings UM, Paulis L, Unger T, and Bader M. (2011) Emerging drugs which target the renin-angiotensin-aldosterone system. Expert Opin Emerg Drugs 16: 619-630. [Crossref]
- Silva DM, Vianna HR, Cortes SF, Campagnole-Santos MJ, Santos RA, et al. (2007) Evidence for a new angiotensin-(1-7) receptor subtype in the aorta of Sprague-Dawley rats. Peptides 28: 702-707. [Crossref]
- Castro CH, Santos RA, Ferreira AJ, Bader M, Alenina N, et al. (2005) Evidence for a functional interaction of the angiotensin-(1-7) receptor Mas with AT1 and AT2 receptors in the mouse heart. Hypertension 46: 937-942. [Crossref]
- Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, et al. (2005) G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111: 1806-1813. [Crossref]
- Canals M, Jenkins L, Kellett E, Milligan G (2006) Up-regulation of the angiotensin II type 1 receptor by the MAS proto-oncogene is due to constitutive activation of Gq/G11 by MAS. J Biol Chem 281: 16757-16767. [Crossref]
- Bosnyak S, Jones ES, Christopoulos A, Aguilar MI, Thomas WG, et al. (2011) Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci (Lond) 121: 297-303. [Crossref]
- Santos RA, Campagnole-Santos MJ, Andrade SP (2000) Angiotensin-(1-7): an update. Regul Pept 91: 45-62. [Crossref]
- Ferrario CM, Chappell MC, Dean RH, Iyer SN (1998) Novel angiotensin peptides regulate blood pressure, endothelial function, and natriuresis. J Am Soc Nephrol 9: 1716-1722. [Crossref]
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, et al. (2002) Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277: 14838-14843. [Crossref]
- Allred AJ, Diz DI, Ferrario CM, Chappell MC (2000) Pathways for angiotensin-(1---7) metabolism in pulmonary and renal tissues. Am J Physiol Renal Physiol 279: F841-850. [Crossref]
- Chappell MC, Pirro NT, Sykes A, Ferrario CM (1998) Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 31: 362-367. [Crossref]
- Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, et al. (1998) N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1-7) and keto-ACE. Hypertension 31: 912-917. [Crossref]
- Yamada K, Iyer SN, Chappell MC, Ganten D, Ferrario CM (1998) Converting enzyme determines plasma clearance of angiotensin-(1-7). Hypertension 32: 496-502. [Crossref]
- Gembardt F, Sterner-Kock A, Imboden H, Spalteholz M, Reibitz F, et al. (2005) Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 26: 1270-1277. [Crossref]
- Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, et al. (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203: 631-637. [Crossref]
- Komatsu T, Suzuki Y, Imai J, Sugano S, Hida M, et al. (2002) Molecular cloning, mRNA expression and chromosomal localization of mouse angiotensin-converting enzyme-related carboxypeptidase (mACE2). DNA Seq 13: 217-220. [Crossref]
- Li N, Zimpelmann J, Cheng K, Wilkins JA, Burns KD (2005) The role of angiotensin converting enzyme 2 in the generation of angiotensin 1-7 by rat proximal tubules. Am J Physiol Renal Physiol 288: F353-362. [Crossref]
- DelliPizzi AM, Hilchey SD, Bell-Quilley CP (1994) Natriuretic action of angiotensin(1-7). Br J Pharmacol 111: 1-3. [Crossref]
- Vallon V, Heyne N, Richter K, Khosla MC, Fechter K (1998) [7-D-ALA]-angiotensin 1-7 blocks renal actions of angiotensin 1-7 in the anesthetized rat. J Cardiovasc Pharmacol 32: 164-167. [Crossref]
- Chappell MC, Modrall JG, Diz DI, Ferrario CM (2004) Novel aspects of the renal renin-angiotensin system: angiotensin-(1-7), ACE2 and blood pressure regulation. Contrib Nephrol 143: 77-89. [Crossref]
- Handa RK, Ferrario CM, Strandhoy JW (1996) Renal actions of angiotensin-(1-7): in vivo and in vitro studies. Am J Physiol 270: F141-147. [Crossref]
- Bürgelová M, Kramer HJ, Teplan V, Velicková G, Vítko S, et al. (2002) Intrarenal infusion of angiotensin-(1-7) modulates renal functional responses to exogenous angiotensin II in the rat. Kidney Blood Press Res 25: 202-210. [Crossref]
- Simões e Silva AC, Bello AP, Baracho NC, Khosla MC, Santos RA (1998) Diuresis and natriuresis produced by long term administration of a selective Angiotensin-(1-7) antagonist in normotensive and hypertensive rats. Regul Pept 74: 177-184. [Crossref]
- Santos RA, Simões e Silva AC, Magaldi AJ, Khosla MC, Cesar KR, et al. (1996) Evidence for a physiological role of angiotensin-(1-7) in the control of hydroelectrolyte balance. Hypertension 27: 875-884. [Crossref]
- Joyner J, Neves L, Ferrario C, Brosnihan K (2008) Administration of D-Alanine-[Ang-(1-7)] (A-779) Prior to Pregnancy in Sprague Dawley Rats Produces Antidiuresis in Late Gestation. J Am Soc Hypertens 2: 425-430. [Crossref]
- Joyner J, Neves LA, Stovall K, Ferrario CM, Brosnihan KB (2008) Angiotensin-(1-7) serves as an aquaretic by increasing water intake and diuresis in association with downregulation of aquaporin-1 during pregnancy in rats. Am J Physiol Regul Integr Comp Physiol 294: R1073-1080. [Crossref]
- Magaldi AJ, Cesar KR, de Araújo M, Simões e Silva AC, Santos RA (2003) Angiotensin-(1-7) stimulates water transport in rat inner medullary collecting duct: evidence for involvement of vasopressin V2 receptors. Pflugers Arch 447: 223-230. [Crossref]
- Heller J, Kramer HJ, Malý J, Cervenka L, Horácek V (2000) Effect of intrarenal infusion of angiotensin-(1-7) in the dog. Kidney Blood Press Res 23: 89-94. [Crossref]
- Garcia NH, Garvin JL (1994) Angiotensin 1-7 has a biphasic effect on fluid absorption in the proximal straight tubule. J Am Soc Nephrol 5: 1133-1138. [Crossref]
- Andreatta-van Leyen S, Romero MF, Khosla MC, Douglas JG (1993) Modulation of phospholipase A2 activity and sodium transport by angiotensin-(1-7). Kidney Int 44: 932-936. [Crossref]
- Caruso-Neves C, Lara LS, Rangel LB, Grossi AL, and Lopes AG. Angiotensin-(1-7) modulates the ouabain-insensitive Na+-ATPase activity from basolateral membrane of the proximal tubule. Biochim Biophys Acta 1467: 189-197, 2000.
- De Souza AM, Lopes AG, Pizzino CP, Fossari RN, Miguel NC, et al. (2004) Angiotensin II and angiotensin-(1-7) inhibit the inner cortex Na+ -ATPase activity through AT2 receptor. Regul Pept 120: 167-175. [Crossref]
- Hilchey SD, Bell-Quilley CP (1995) Association between the natriuretic action of angiotensin-(1-7) and selective stimulation of renal prostaglandin I2 release. Hypertension 25: 1238-1244. [Crossref]
- Moe OW (1999) Acute regulation of proximal tubule apical membrane Na/H exchanger NHE-3: role of phosphorylation, protein trafficking, and regulatory factors. J Am Soc Nephrol 10: 2412-2425. [Crossref]
- Joyner J, Neves LA, Granger JP, Alexander BT, Merrill DC, et al. (2007) Temporal-spatial expression of ANG-(1-7) and angiotensin-converting enzyme 2 in the kidney of normal and hypertensive pregnant rats. Am J Physiol Regul Integr Comp Physiol 293: R169-177. [Crossref]
- Pendergrass KD, Pirro NT, Westwood BM, Ferrario CM, Brosnihan KB, et al. (2008) Sex differences in circulating and renal angiotensins of hypertensive mRen(2). Lewis but not normotensive Lewis rats. Am J Physiol Heart Circ Physiol 295: H10-20. [Crossref]
- Castelo-Branco RC, Leite-Delova DC, and de Mello-Aires M. (2013) Dose-dependent effects of angiotensin-(1-7) on the NHE3 exchanger and [Ca(2+)](i) in in vivo proximal tubules. Am J Physiol Renal Physiol 304: F1258-1265. [Crossref]
- Casavola V, Guerra L, Helmle-Kolb C, Reshkin SJ, Murer H (1992) Na+/H(+)-exchange in A6 cells: polarity and vasopressin regulation. J Membr Biol 130: 105-114. [Crossref]
- Alenina N, Xu P, Rentzsch B, Patkin EL, Bader M (2008) Genetically altered animal models for Mas and angiotensin-(1-7). Exp Physiol 93: 528-537. [Crossref]
- da Silveira KD, Pompermayer Bosco KS, Diniz LR, Carmona AK, Cassali GD, et al. (2010) ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin Sci (Lond) 119: 385-394. [Crossref]
- Ferrario CM, Varagic J (2010) The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Renal Physiol 298: F1297-1305. [Crossref]
- Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, et al. (2010) Nuclear angiotensin-(1-7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol 299: F983-990. [Crossref]
- Ferrario CM. (2010) New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension 55: 445-452. [Crossref]
- Musa-Aziz R, Oliveira-Souza M, Mello-Aires M (2005) Signaling pathways in the biphasic effect of ANG II on Na+/H+ exchanger in T84 cells. J Membr Biol 205: 49-60. [Crossref]
- Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI (1997) Counterregulatory actions of angiotensin-(1-7). Hypertension 30: 535-541. [Crossref]
- Brown NJ (2013) Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol 9: 459-469. [Crossref]
- Takeda Y, Miyamori I, Yoneda T, Hatakeyama H, Inaba S, et al. (1996) Regulation of aldosterone synthase in human vascular endothelial cells by angiotensin II and adrenocorticotropin. J Clin Endocrinol Metab 81: 2797-2800. [Crossref]
- Takeda Y, Miyamori I, Yoneda T, Iki K, Hatakeyama H, et al. (1994) Synthesis of corticosterone in the vascular wall. Endocrinology 135: 2283-2286. [Crossref]
- Müller J (1987) Regulation of aldosterone biosynthesis. Physiological and clinical aspects. Monogr Endocrinol 29: 1-364. [Crossref]
- Quinn SJ, Williams GH (1988) Regulation of aldosterone secretion. Annu Rev Physiol 50: 409-426. [Crossref]
- Epstein M (2001) Aldosterone as a determinant of cardiovascular and renal dysfunction. J R Soc Med 94: 378-383. [Crossref]
- Spät A, Hunyady L (2004) Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 84: 489-539. [Crossref]
- Hackenthal E, Paul M, Ganten D, Taugner R (1990) Morphology, physiology, and molecular biology of renin secretion. Physiol Rev 70: 1067-1116. [Crossref]
- Kurtz A, Wagner C (1999) Regulation of renin secretion by angiotensin II-AT1 receptors. J Am Soc Nephrol 10 Suppl 11: S162-168. [Crossref]
- Coll AP, Challis BG, Yeo GS, Snell K, Piper SJ, et al. (2004) The effects of proopiomelanocortin deficiency on murine adrenal development and responsiveness to adrenocorticotropin. Endocrinology 145: 4721-4727. [Crossref]
- Cooke BA (1999) Signal transduction involving cyclic AMP-dependent and cyclic AMP-independent mechanisms in the control of steroidogenesis. Mol Cell Endocrinol 151: 25-35. [Crossref]
- Cozza EN, Vila MC, Acevedo-Duncan M, Farese RV, and Gómez-Sánchez CE. (1990) Treatment of primary cultures of calf adrenal glomerulosa cells with adrenocorticotropin (ACTH) and phorbol esters: a comparative study of the effects on aldosterone production and ACTH signaling system. Endocrinology 126: 2169-2176. [Crossref]
- Krug AW, Grossmann C, Schuster C, Freudinger R, Mildenberger S, et al. (2003) Aldosterone stimulates epidermal growth factor receptor expression. J Biol Chem 278: 43060-43066. [Crossref]
- Moghal N, Sternberg PW (1999) Multiple positive and negative regulators of signaling by the EGF-receptor. Curr Opin Cell Biol 11: 190-196. [Crossref]
- Wada T, Penninger JM (2004) Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23: 2838-2849. [Crossref]
- Cowan KJ, Storey KB (2003) Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol 206: 1107-1115. [Crossref]
- Michlig S, Mercier A, Doucet A, Schild L, Horisberger JD, Rossier BC, and Firsov D. ERK1/2 controls Na,K-ATPase activity and transepithelial sodium transport in the principal cell of the cortical collecting duct of the mouse kidney. J Biol Chem 279: 51002-51012, 2004.
- Brem AS, Gong R (2015) Therapeutic targeting of aldosterone: a novel approach to the treatment of glomerular disease. Clin Sci (Lond) 128: 527-535. [Crossref]
- Guichard JL, Clark D 3rd, Calhoun DA, Ahmed MI (2013) Aldosterone receptor antagonists: current perspectives and therapies. Vasc Health Risk Manag 9: 321-331. [Crossref]
- Mathew JT, Patni H, Chaudhary AN, Liang W, Gupta A, et al. (2008) Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol Renal Physiol 295: F73-81. [Crossref]
- Morgado-Pascual JL, Rayego-Mateos S, Valdivielso JM, Ortiz A, Egido J, et al. (2015) Paricalcitol Inhibits Aldosterone-Induced Proinflammatory Factors by Modulating Epidermal Growth Factor Receptor Pathway in Cultured Tubular Epithelial Cells. Biomed Res Int 2015: 783538. [Crossref]
- Rafiq K, Hitomi H, Nakano D, Nishiyama A (2011) Pathophysiological roles of aldosterone and mineralocorticoid receptor in the kidney. J Pharmacol Sci 115: 1-7. [Crossref]
- Kitada K, Nakano D, Hitomi H, Kobori H, Deguchi K, et al. (2012) Aldosterone induces p21-regulated apoptosis via increased synthesis and secretion of tumour necrosis factor-a in human proximal tubular cells. Clin Exp Pharmacol Physiol 39: 858-863. [Crossref]
- Liang W, Chen C, Shi J, Ren Z, Hu F, et al. (2011) Disparate effects of eplerenone, amlodipine and telmisartan on podocyte injury in aldosterone-infused rats. Nephrol Dial Transplant 26: 789-799. [Crossref]
- Briet M, Schiffrin EL (2013) Vascular actions of aldosterone. J Vasc Res 50: 89-99. [Crossref]
- Connell JM, Davies E (2005) The new biology of aldosterone. J Endocrinol 186: 1-20. [Crossref]
- Cho JH, Musch MW, Bookstein CM, McSwine RL, Rabenau K, et al. (1998) Aldosterone stimulates intestinal Na+ absorption in rats by increasing NHE3 expression of the proximal colon. Am J Physiol 274: C586-594. [Crossref]
- Donowitz M, De La Horra C, Calonge ML, Wood IS, Dyer J, et al. (1998) In birds, NHE2 is major brush-border Na+/H+ exchanger in colon and is increased by a low-NaCl diet. Am J Physiol 274: R1659-1669. [Crossref]
- Pergher PS, Leite-Dellova D, de Mello-Aires M (2009) Direct action of aldosterone on bicarbonate reabsorption in in vivo cortical proximal tubule. Am J Physiol Renal Physiol 296: F1185-1193. [Crossref]
- Braga-Sobrinho C, Leite-Dellova DC, Mello-Aires M (2012) Action of ANP on the nongenomic dose-dependent biphasic effect of aldosterone on NHE1 in proximal S3 segment. J Steroid Biochem Mol Biol 128: 89-97. [Crossref]
- Leite-Dellova DC, Oliveira-Souza M, Malnic G, Mello-Aires M (2008) Genomic and nongenomic dose-dependent biphasic effect of aldosterone on Na+/H+ exchanger in proximal S3 segment: role of cytosolic calcium. Am J Physiol Renal Physiol 295: F1342-1352. [Crossref]
- Salyer SA, Parks J, Barati MT, Lederer ED, Clark BJ, et al. (2013) Aldosterone regulates Na(+), K(+) ATPase activity in human renal proximal tubule cells through mineralocorticoid receptor. Biochim Biophys Acta 1833: 2143-2152. [Crossref]
- Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, et al. (1998) The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A 95: 14552-14557. [Crossref]
- Leite-Dellova DC, Malnic G, Mello-Aires M (2011) Genomic and nongenomic stimulatory effect of aldosterone on H+-ATPase in proximal S3 segments. Am J Physiol Renal Physiol 300: F682-691. [Crossref]
- Náray-Fejes-Tóth A, Helms MN, Stokes JB, Fejes-Tóth G (2004) Regulation of sodium transport in mammalian collecting duct cells by aldosterone-induced kinase, SGK1: structure/function studies. Mol Cell Endocrinol 217: 197-202. [Crossref]
- Stockand JD (2002) New ideas about aldosterone signaling in epithelia. Am J Physiol Renal Physiol 282: F559-576. [Crossref]
- Snyder PM, Olson DR, Thomas BC (2002) Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277: 5-8. [Crossref]
- Mastroberardino L, Spindler B, Forster I, Loffing J, Assandri R, et al. (1998) Ras pathway activates epithelial Na+ channel and decreases its surface expression in Xenopus oocytes. Mol Biol Cell 9: 3417-3427. [Crossref]
- Stockand JD, Meszaros JG (2003) Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol 284: H176-184. [Crossref]
- Blazer-Yost BL, Liu X, Helman SI (1998) Hormonal regulation of ENaCs: insulin and aldosterone. Am J Physiol 274: C1373-1379. [Crossref]
- Geering K, Béguin P, Garty H, Karlish S, Füzesi M, et al. (2003) FXYD proteins: new tissue- and isoform-specific regulators of Na,K-ATPase. Ann N Y Acad Sci 986: 388-394. [Crossref]
- Aizman R, Asher C, Füzesi M, Latter H, Lonai P, et al. (2002) Generation and phenotypic analysis of CHIF knockout mice. Am J Physiol Renal Physiol 283: F569-577. [Crossref]
- Garty H, Lindzen M, Füzesi M, Aizman R, Goldshleger R, et al. (2003) A specific functional interaction between CHIF and Na,K-ATPase: role of FXYD proteins in the cellular regulation of the pump. Ann N Y Acad Sci 986: 395-400. [Crossref]
- SIMPSON SA, TAIT JF, WETTSTEIN A, NEHER R, VON EUW J, et al. (1953) [Isolation from the adrenals of a new crystalline hormone with especially high effectiveness on mineral metabolism]. Experientia 9: 333-335. [Crossref]
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, et al. (1987) Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237: 268-275. [Crossref]
- Rogerson FM, Yao Y, Smith BJ, and Fuller PJ. (2004) Differences in the determinants of eplerenone, spironolactone and aldosterone binding to the mineralocorticoid receptor. Clin Exp Pharmacol Physiol 31: 704-709. [Crossref]
- Karin M (1998) New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell 93: 487-490. [Crossref]
- De Kloet ER, Ratka A, Reul JM, Sutanto W, Van Eekelen JA (1987) Corticosteroid receptor types in brain: regulation and putative function. Ann N Y Acad Sci 512: 351-361. [Crossref]
- Gómez-Sánchez EP (1997) Central hypertensive effects of aldosterone. Front Neuroendocrinol 18: 440-462. [Crossref]
- Karssen AM, Meijer OC, van der Sandt IC, Lucassen PJ, de Lange EC, et al. (2001) Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology 142: 2686-2694. [Crossref]
- Ueda K, Okamura N, Hirai M, Tanigawara Y, Saeki T, et al. (1992) Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J Biol Chem 267: 24248-24252. [Crossref]
- Montezano AC, Callera GE, Yogi A, He Y, Tostes RC, et al. (2008) Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Src-regulated redox-sensitive RhoA pathways. Arterioscler Thromb Vasc Biol 28: 1511-1518. [Crossref]
- Briet M, Schiffrin EL (2010) Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol 6: 261-273. [Crossref]
- Sakai RR, Ma LY, Zhang DM, McEwen BS, and Fluharty SJ. (1996) Intracerebral administration of mineralocorticoid receptor antisense oligonucleotides attenuate adrenal steroid-induced salt appetite in rats. Neuroendocrinology 64: 425-429. [Crossref]
- Christ M, Sippe K, Eisen C, Wehling M (1994) Non-classical receptors for aldosterone in plasma membranes from pig kidneys. Mol Cell Endocrinol 99: R31-34. [Crossref]
- Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M (2000) Multiple actions of steroid hormones--a focus on rapid, nongenomic effects. Pharmacol Rev 52: 513-556. [Crossref]
- Haseroth K, Gerdes D, Berger S, Feuring M, Günther A, et al. (1999) Rapid nongenomic effects of aldosterone in mineralocorticoid-receptor-knockout mice. Biochem Biophys Res Commun 266: 257-261. [Crossref]
- Ozegović B, Dobrović-Jenik D, Milković S (1988) Solubilization of rat kidney plasma membrane proteins associated with 3H-aldosterone. Exp Clin Endocrinol 92: 194-198. [Crossref]
- A-Awqati Q, Norby LH, Mueller A, Steinmetz PR (1976) Characteristics of stimulation of H+ transport by aldosterone in turtle urinary bladder. J Clin Invest 58: 351-358. [Crossref]
- Harvey BJ (1992) Energization of sodium absorption by the H(+)-ATPase pump in mitochondria-rich cells of frog skin. J Exp Biol 172: 289-309. [Crossref]
- Wehling M, Ulsenheimer A, Schneider M, Neylon C, and Christ M. Rapid effects of aldosterone on free intracellular calcium in vascular smooth muscle and endothelial cells: subcellular localization of calcium elevations by single cell imaging. Biochem Biophys Res Commun 204: 475-481, 1994
- Estrada M, Liberona JL, Miranda M, Jaimovich E (2000) Aldosterone- and testosterone-mediated intracellular calcium response in skeletal muscle cell cultures. Am J Physiol Endocrinol Metab 279: E132-139. [Crossref]
- Christ M, Eisen C, Aktas J, Theisen K, and Wehling M. (1993) The inositol-1,4,5-trisphosphate system is involved in rapid effects of aldosterone in human mononuclear leukocytes. J Clin Endocrinol Metab 77: 1452-1457. [Crossref]
- Sato A, Liu JP, Funder JW (1997) Aldosterone rapidly represses protein kinase C activity in neonatal rat cardiomyocytes in vitro. Endocrinology 138: 3410-3416. [Crossref]
- Doolan CM, Harvey BJ (1996) Rapid effects of steroid hormones on free intracellular calcium in T84 colonic epithelial cells. Am J Physiol 271: C1935-1941. [Crossref]
- Winter DC, Schneider MF, O'Sullivan GC, Harvey BJ, Geibel JP (1999) Rapid effects of aldosterone on sodium-hydrogen exchange in isolated colonic crypts. J Membr Biol 170: 17-26. [Crossref]
- Gekle M, Silbernagl S, Oberleithner H (1997) The mineralocorticoid aldosterone activates a proton conductance in cultured kidney cells. Am J Physiol 273: C1673-1678. [Crossref]
- Zhou ZH, Bubien JK (2001) Nongenomic regulation of ENaC by aldosterone. Am J Physiol Cell Physiol 281: C1118-1130. [Crossref]
- Maguire D, MacNamara B, Cuffe JE, Winter D, Doolan CM, et al. (1999) Rapid responses to aldosterone in human distal colon. Steroids 64: 51-63. [Crossref]
- Good DW, George T, Watts BA 3rd (2003) Aldosterone potentiates 1,25-dihydroxyvitamin D3 action in renal thick ascending limb via a nongenomic, ERK-dependent pathway. Am J Physiol Cell Physiol 285: C1122-1130. [Crossref]
- Le Moëllic C, Ouvrard-Pascaud A, Capurro C, Cluzeaud F, Fay M, et al. (2004) Early nongenomic events in aldosterone action in renal collecting duct cells: PKCalpha activation, mineralocorticoid receptor phosphorylation, and cross-talk with the genomic response. J Am Soc Nephrol 15: 1145-1160. [Crossref]
- Gekle M, Golenhofen N, Oberleithner H, and Silbernagl S. (1996) Rapid activation of Na+/H+ exchange by aldosterone in renal epithelial cells requires Ca2+ and stimulation of a plasma membrane proton conductance. Proc Natl Acad Sci U S A 93: 10500-10504. [Crossref]
- Good DW, George T, Watts BA 3rd (2006) Nongenomic regulation by aldosterone of the epithelial NHE3 Na(+)/H(+) exchanger. Am J Physiol Cell Physiol 290: C757-763. [Crossref]
- Winter C, Schulz N, Giebisch G, Geibel JP, Wagner CA (2004) Nongenomic stimulation of vacuolar H+-ATPases in intercalated renal tubule cells by aldosterone. Proc Natl Acad Sci U S A 101: 2636-2641. [Crossref]
- Grossmann C, Gekle M (2009) New aspects of rapid aldosterone signaling. Mol Cell Endocrinol 308: 53-62. [Crossref]
- 275.Thomas W, Harvey BJ (2011) Mechanisms underlying rapid aldosterone effects in the kidney. Annu Rev Physiol 73: 335-357. [Crossref]
- Wildling L, Hinterdorfer P, Kusche-Vihrog K, Treffner Y, and Oberleithner H. (2009) Aldosterone receptor sites on plasma membrane of human vascular endothelium detected by a mechanical nanosensor. Pflugers Arch 458: 223-230. [Crossref]Norman AW, Mizwicki MT, Norman DP (2004) Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov 3: 27-41. [Crossref]
- Gekle M, Freudinger R, Mildenberger S, Silbernagl S (2002) Rapid actions of aldosterone on cells from renal epithelium: the possible role of EGF-receptor signaling. Steroids 67: 499-504. [Crossref]
- Coutinho P, Vega C, Pojoga LH, Rivera A, Prado GN, et al. (2014) Aldosterone's rapid, nongenomic effects are mediated by striatin: a modulator of aldosterone's effect on estrogen action. Endocrinology 155: 2233-2243. [Crossref]
- Booth RE, Johnson JP, Stockand JD (2002) Aldosterone. Adv Physiol Educ 26: 8-20. [Crossref]
- Ebata S, Muto S, Okada K, Nemoto J, Amemiya M, et al. (1999) Aldosterone activates Na+/H+ exchange in vascular smooth muscle cells by nongenomic and genomic mechanisms. Kidney Int 56: 1400-1412. [Crossref]
- Oberleithner H, Weigt M, Westphale HJ, Wang W (1987) Aldosterone activates Na+/H+ exchange and raises cytoplasmic pH in target cells of the amphibian kidney. Proc Natl Acad Sci U S A 84: 1464-1468. [Crossref]
- Watts BA 3rd, George T, Good DW (2006) Aldosterone inhibits apical NHE3 and HCO3- absorption via a nongenomic ERK-dependent pathway in medullary thick ascending limb. Am J Physiol Renal Physiol 291: F1005-1013. [Crossref]
- Schäfer C, Shahin V, Albermann L, Schillers H, Hug MJ, et al. (2003) Intracellular calcium: a prerequisite for aldosterone action. J Membr Biol 196: 157-162. [Crossref]
- Mavani GP, DeVita MV, Michelis MF (2015) A review of the nonpressor and nonantidiuretic actions of the hormone vasopressin. Front Med (Lausanne) 2: 19. [Crossref]
- Bankir L (2001) Antidiuretic action of vasopressin: quantitative aspects and interaction between V1a and V2 receptor-mediated effects. Cardiovasc Res 51: 372-390. [Crossref]
- Morgenthaler NG, Struck J, Alonso C, Bergmann A (2006) Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem 52: 112-119. [Crossref]
- VERNEY EB (1947) The antidiuretic hormone and the factors which determine its release. Proc R Soc Lond B Biol Sci 135: 25-106. [Crossref]
- Danziger J, Zeidel ML (2015) Osmotic homeostasis. Clin J Am Soc Nephrol 10: 852-862. [Crossref]
- Oliet SH, Bourque CW (1993) Mechanosensitive channels transduce osmosensitivity in supraoptic neurons. Nature 364: 341-343. [Crossref]
- Schrier RW, Berl T, Anderson RJ (1979) Osmotic and nonosmotic control of vasopressin release. Am J Physiol 236: F321-332. [Crossref]
- Kannan H, and Yagi K. (1978) Supraoptic neurosecretory neurons: evidence for the existence of coverging intpus both from carotid baroreceptors and osmoreceptors. Brain Res 145: 385-390. [Crossref]
- Ishikawa S, Saito T, and Yoshida S. (1980) The effect of osmotic pressure and angiotensin II on arginine vasopressin release from guinea pig hypothalamo-neurohypophyseal complex in organ culture. Endocrinology 106: 1571-1578. [Crossref]
- Satoh K, Oti T, Katoh A, Ueta Y, Morris JF, et al. (2015) In vivo processing and release into the circulation of GFP fusion protein in arginine vasopressin enhanced GFP transgenic rats: response to osmotic stimulation. FEBS J 282: 2488-2499. [Crossref]
- Sved AF, Imaizumi T, Talman WT, and Reis DJ. (1985) Vasopressin contributes to hypertension caused by nucleus tractus solitarius lesions. Hypertension 7: 262-267. [Crossref]
- Schrier RW, Berl T (1973) Mechanism of effect of alpha adrenergic stimulation with norepinephrine on renal water excretion. J Clin Invest 52: 502-511. [Crossref]
- Schrier RW, Berl T (1972) Mechanism of the antidiuretic effect associated with interruption of parasympathetic pathways. J Clin Invest 51: 2613-2620. [Crossref]
- Robertson GL (1976) The regulation of vasopressin function in health and disease. Recent Prog Horm Res 33: 333-385. [Crossref]
- Dunn FL, Brennan TJ, Nelson AE, Robertson GL (1973) The role of blood osmolality and volume in regulating vasopressin secretion in the rat. J Clin Invest 52: 3212-3219. [Crossref]
- Creager MA, Faxon DP, Cutler SS, Kohlmann O, Ryan TJ, et al. (1986) Contribution of vasopressin to vasoconstriction in patients with congestive heart failure: comparison with the renin-angiotensin system and the sympathetic nervous system. J Am Coll Cardiol 7: 758-765. [Crossref]
- Robertson GL, Aycinena P, Zerbe RL (1982) Neurogenic disorders of osmoregulation. Am J Med 72: 339-353. [Crossref]
- Kam PC, Williams S, Yoong FF (2004) Vasopressin and terlipressin: pharmacology and its clinical relevance. Anaesthesia 59: 993-1001. [Crossref]
- Aoyagi T, Izumi Y, Hiroyama M, Matsuzaki T, Yasuoka Y, et al. (2008) Vasopressin regulates the renin-angiotensin-aldosterone system via V1a receptors in macula densa cells. Am J Physiol Renal Physiol 295: F100-107. [Crossref]
- Arpin-Bott MP, Kaissling B, Waltisperger E, Rabhi M, Saussine P, et al. (2002) Historadioautographic localization of oxytocin and V1a vasopressin binding sites in the kidney of developing and adult rabbit, mouse and merione and of adult human. Exp Nephrol 10: 196-208. [Crossref]
- Park F, Mattson DL, Skelton MM, Cowley AW Jr (1997) Localization of the vasopressin V1a and V2 receptors within the renal cortical and medullary circulation. Am J Physiol 273: R243-251. [Crossref]
- Verbrugge FH, Steels P, Grieten L, Nijst P, Tang WH, et al. (2015) Hyponatremia in acute decompensated heart failure: depletion versus dilution. J Am Coll Cardiol 65: 480-492. [Crossref]
- Birnbaumer M (2000) Vasopressin receptors. Trends Endocrinol Metab 11: 406-410. [Crossref]
- Lee CR, Watkins ML, Patterson JH, Gattis W, O'connor CM, et al. (2003) Vasopressin: a new target for the treatment of heart failure. Am Heart J 146: 9-18. [Crossref]
- Lolait SJ, O'Carroll AM, Mahan LC, Felder CC, Button DC, et al. (1995) Extrapituitary expression of the rat V1b vasopressin receptor gene. Proc Natl Acad Sci U S A 92: 6783-6787. [Crossref]
- Monstein HJ, Truedsson M, Ryberg A, Ohlsson B (2008) Vasopressin receptor mRNA expression in the human gastrointestinal tract. Eur Surg Res 40: 34-40. [Crossref]
- Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl W, et al. (2007) Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol 293: F1166-1177. [Crossref]
- Ishikawa SE. (2015) Hyponatremia Associated with Heart Failure: Pathological Role of Vasopressin-Dependent Impaired Water Excretion. J Clin Med 4: 933-947. [Crossref]
- Lolait SJ, O'Carroll AM, McBride OW, Konig M, Morel A, et al. (1992) Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 357: 336-339. [Crossref]
- 314. Fenton RA, Knepper MA (2007) Mouse models and the urinary concentrating mechanism in the new millennium. Physiol Rev 87: 1083-1112. [Crossref]
- Reif MC, Troutman SL, Schafer JA (1986) Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J Clin Invest 77: 1291-1298. [Crossref]
- Tomita K, Pisano JJ, and Knepper MA. (1985) Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest 76: 132-136. [Crossref]
- Snyder PM (2005) Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology 146: 5079-5085. [Crossref]
- Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, et al. (2000) Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol 279: F46-53. [Crossref]
- Capasso G, Malnic G, Wang T, Giebisch G (1994) Acidification in mammalian cortical distal tubule. Kidney Int 45: 1543-1554. [Crossref]
- Handler JS, Orloff J (1981) Antidiuretic hormone. Annu Rev Physiol 43: 611-624. [Crossref]
- Bichara M, Mercier O, Houillier P, Paillard M, Leviel F (1987) Effects of antidiuretic hormone on urinary acidification and on tubular handling of bicarbonate in the rat. J Clin Invest 80: 621-630. [Crossref]
- Good DW. (1990) Inhibition of bicarbonate absorption by peptide hormones and cyclic adenosine monophosphate in rat medullary thick ascending limb. J Clin Invest 85: 1006-1013. [Crossref]
- Tomita K, Pisano JJ, Burg MB, Knepper MA (1986) Effects of vasopressin and bradykinin on anion transport by the rat cortical collecting duct. Evidence for an electroneutral sodium chloride transport pathway. J Clin Invest 77: 136-141. [Crossref]
- Sun AM, Kikeri D, Hebert SC (1992) Vasopressin regulates apical and basolateral Na(+)-H+ antiporters in mouse medullary thick ascending limbs. Am J Physiol 262: F241-247. [Crossref]
- Moses AM, Steciak E (1986) Urinary and metabolic clearances of arginine vasopressin in normal subjects. Am J Physiol 251: R365-370. [Crossref]
- Ando Y, Tabei K, Asano Y (1991) Luminal vasopressin modulates transport in the rabbit cortical collecting duct. J Clin Invest 88: 952-959. [Crossref]
- Naruse M, Yoshitomi K, Hanaoka K, Imai M, Kurokawa K (1995) Electrophysiological study of luminal and basolateral vasopressin in rabbit cortical collecting duct. Am J Physiol 268: F20-29. [Crossref]
- Burgess WJ, Balment RJ, Beck JS (1994) Effects of luminal vasopressin on intracellular calcium in microperfused rat medullary thick ascending limb. Ren Physiol Biochem 17: 1-9. [Crossref]
- Ikeda M, Yoshitomi K, Imai M, Kurokawa K (1994) Cell Ca2+ response to luminal vasopressin in cortical collecting tubule principal cells. Kidney Int 45: 811-816. [Crossref]
- Harvey N, Jones JJ, Lee J (1967) The renal clearance and plasma binding of vasopressin in the dog. J Endocrinol 38: 163-171. [Crossref]
- Kimura T, Share L (1981) Characterization of the renal handling of vasopressin in the dog by stop-flow analysis. Endocrinology 109: 2089-2094. [Crossref]
- GINSBURG M (1957) The clearance of vasopressin from the splanchnic vascular area and the kidneys. J Endocrinol 16: 217-226. [Crossref]
- Borensztein P, Juvin P, Vernimmen C, Poggioli J, Paillard M, et al. (1993) cAMP-dependent control of Na+/H+ antiport by AVP, PTH, and PGE2 in rat medullary thick ascending limb cells. Am J Physiol 264: F354-364. [Crossref]
- Grider J, Falcone J, Kilpatrick E, Ott C, Jackson B (1996) Effect of luminal vasopressin on NaCl transport in the medullary thick ascending limb of the rat. Eur J Pharmacol 313: 115-118. [Crossref]
- Oliveira-Souza M, and Mello-Aires M. (2001) Effect of arginine vasopressin and ANP on intracellular pH and cytosolic free [Ca2+] regulation in MDCK cells. Kidney Int 60: 1800-1808. [Crossref]
- Musa-Aziz R, Barreto-Chaves ML, De Mello-Aires M (2002) Peritubular AVP regulates bicarbonate reabsorption in cortical distal tubule via V(1) and V(2) receptors. Am J Physiol Renal Physiol 282: F256-264. [Crossref]
- Ando Y, Asano Y (1993) Functional evidence for an apical V1 receptor in rabbit cortical collecting duct. Am J Physiol 264: F467-471. [Crossref]
- Berl T, and Robertson GL. Pathophysiology of water methabolism. In: The Kidney, edited by style="font-size:12.0pt slE-U, line-height:, 115%, font-family:"Times New Roman" s, Roman" m-f-f-fTN, mso-ansi-language:EN-US, mso-fareast-language:PT-BR, mso-bidi-language:AR-SA, and mso-bidi-font-style:italic">Brenner BM eBaRs. Phyladelphia: 6th Ed. Saunders 2000.
- Santos RA, Baracho NC (1992) Angiotensin-(1-7) is a potent antidiuretic peptide in rats. Braz J Med Biol Res 25: 651-654. [Crossref]
- AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U (2001) The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem 276: 39721-39726. [Crossref]
- Carraro-Lacroix LR, Girardi AC, Malnic G (2009) Long-term regulation of vacuolar H(+)-ATPase by angiotensin II in proximal tubule cells. Pflugers Arch 458: 969-979. [Crossref]
- Levine DZ, Iacovitti M, Buckman S, Harrison V (1994) In vivo modulation of rat distal tubule net HCO3 flux by VIP, isoproterenol, angiotensin II, and ADH. Am J Physiol 266: F878-883. [Crossref]