Abstract
The increase in microarray usage has led to a parallel increase in the discoveries of new genotypes and phenotypes that are partially similar to known genetic syndromes. OculoDentoDigital Dysplasia (ODDD) syndrome is caused by a mutation in the connexin-43 GJA1 gene on the long arm of chromosome 6. We report a 15 year old girl who presents with a partial 12 q monosomy with ODDD-like features including microphthalmia, microcorneas, microdontia, camptodactyly of the fingers and toes, short stature, developmental and speech delays, and fine motor and hand-eye incoordination. Chromosomal Microarray Analysis showed a 12.987 MB loss of in chromosome 12q21.2 to q21.33, and included more than 20 genes, none of which have any known relation to connexin-43. Our patient did not have any deletions or mutations found on chromosome 6q. Two longer deletions on the long arm of chromosome 12 have been documented in connection to the CardioFacioCutaneous (CFC) syndrome, implicating the region 12q21.2-q22 as a candidate region for the gene or genes causing CFC syndrome. This is the first report of a patient with phenotypic features of ODDD syndrome with a microdeletion on chromosome 12q21. Patients who may have an ODDD like phenotype but are negative for connexin-43 mutation should be evaluated for a 12q21 deletion.
Key words
12q deletion, oculodentodigital dysplasia, cardiofaciocutaneous syndrome
Introduction
OculoDentoDigital Dysplasia (ODDD) is an uncommon genetically inherited disease described as having a distinctive facial appearance with variable involvement of the eyes, teeth, and digits. It follows an autosomal dominant inheritance from a mutation in the connexin-43 (GJA1) gene on chromosome 6q. Facial features include narrow pinched nose with a narrow nasal bridge, prominent columella, and thin anteverted nares. Ocular features comprise prominent epicanthal folds, eye abnormalities that can lead to vision loss (myopia), micro-ophthalmia, microcornea, cataracts, and glaucoma. Dental abnormalities consist of microdontia, enamel hypoplasia, and caries. Digit malformations include syndactyly and camptodactyly [1].
Monsomy of the long arm of chromosome 12 is a rare laboratory finding with 11 previous reports. Of these previous studies two relatively recent reports by Raun et al. in 2000 and 2002 implicated the region from 12q21.2 to q22 as a candidate region for the gene or genes causing CardioFacioCutaneous (CFC) syndrome [2,3]. CFC is a genetic disorder characterized by heart defects, facial anomalies, and cutaneous abnormalities, previously reported as typically resulting from a de novo dominant mutation [1].
James et al. in 2005 reported another patient with 12q deletion with non-specific features including developmental delays, microcephaly, facial features including a high nasal bridge, fine hair and sparse facial hair, narrow hands and feet, and a hyperkeratotic rash [4]. In 1989 Watson et al. described an infant with an interstitial deletion from 12q15 to q21.2 with frontal bossing, small sunken eyes, a beaked nose, low-set ears, thin upper lip, high-arched palate, syndactyly of the 2nd and 3rd toes of the left foot, cutis marmorata, and developmental delay [5].
With the increased use and application of microarray analysis there has been an increased incidence of patients presenting with phenotypic characteristics of a certain genetic syndrome only to have the microarray results show a different genotype that are not consistent with the literature. Microarray has also helped to characterize patients that present with a phenotypic combination that has either not been previously documented or documented as an atypical presentation of a similar syndrome that is genotypically different. These increasing situations have paved the way for characterization of the “Like” syndromes such as Noonan-like, Prader-Willi-like, and others.
We present a 15 year old female with OculoDentoDigital Dysplasia-Like phenotype with a partial 12 q monosomy.
Case report
Our patient is a 15 year old female who was brought in by her grandparents because of a previously diagnosed chromosomal abnormality. She was born full term via uncomplicated spontaneous vaginal delivery. The pregnancy was uncomplicated; however, her mother only received prenatal care during the last month of pregnancy. She was evaluated at the age of 4 years because of developmental delays. Grandparents describe her as a healthy young girl with no history of hospitalizations or recurrent infections. She was found to have a deletion of the long arm of chromosome 12.
On our initial evaluation she had a short attention span, speech delays, and minor bilateral myopia (R 20/40; L 20/30). She is in Special Education classes and struggles particularly with math. She is small for her age. Weight was 36.5 kg, and height was 150 cm; both below the 3rd percentile. Also, the frontal occipital circumference was 51 cm which correlates near the 17th percentile. Physical examination also revealed minimal scoliosis, small corneas, small teeth, as well as camptodactyly and partial syndactaly (Figures 1-4).
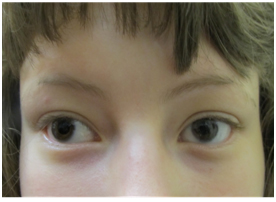
Figure 1. Microcornea
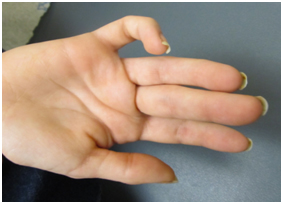
Figure 2. Camptodactyly of fifth digit
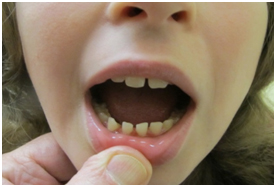
Figure 3. Microdontia
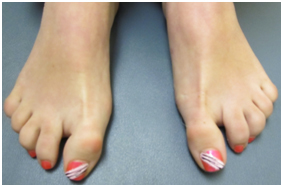
Figure 4. Camptodactyly and Partial syndactaly of 2nd and 3rd toes
Maternal grandparents and mother were all without the abnormalities present in our patient. The patient has two maternal half-brothers, one of which is in the lower percentiles for height and weight, but both are developmentally adequate. The father was described as “normal”, but he was unavailable for follow up history and chromosomal studies.
Methods
Microarray Analysis
Chromosomal microarray analysis (CMA – HR + SNP version 9.1.1) was performed at the Kleberg Cytogenetics Laboratory which revealed a 12.987 Mb deletion on chromosome band 12q21.2q21.33 (figures 5). Two smaller deletions were seen on 11q14.1 and 17p13.1. Two small chromosomal gains were seen on Xp11.4 and Xq13.1. No increased blocks of absence of heterozygosity suggestive of uniparental disomy or consanguinity were detected. None of the smaller deletions or gains have been reported to be associated with a clinical phenotype. Maternal FISH analysis showed no evidence of a rearrangement involving chromosome 12 as seen in our patient.
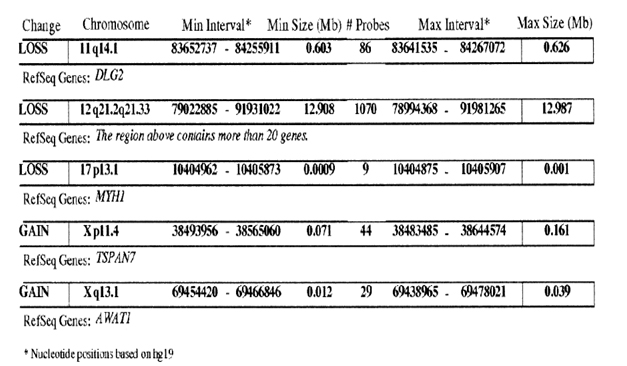
Figure 5. Microarray analysis with 12.987Mb deletion.
MRI showed eye globes that are smaller than average. The total axial length was 21.1 mm for the left eye, and 22 mm for the right eye. According to Bardakjian et al. (2004) this measures as a simple microphthalmia as the lower 2.5% confidence limit for the total axial length is about 21.0 mm [6]. The brain showed no reduction in white matter volume or other white matter abnormalities which is the most common finding in ODDD (Figure 6) [7]. Analysis of chromosome 6 for ODDD mutation was sent to John’s Hopkins DNA Diagnostic Lab showed no evidence of a sequence variation in the coding regions of the GJA1 gene on chromosome 6.
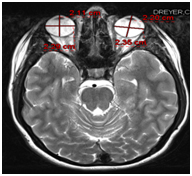
Figure 6. MRI showing small eye globes and negative for white matter abnormalities.

Figure 7. Gene map of deleted region
Discussion
OculoDentoDigital Dysplasia is a syndrome of a constellation of symptoms and phenotypic findings including characteristic facial features of narrow nose and hypoplastic alae nasi, and the previously mentioned abnormalities of the eyes, teeth, and digits of the hands and feet. Other findings include neurologic problems such as developmental delays, dysarthria, neurogenic bladder disturbances, spastic paraperesis, ataxia, anterior tibial muscle weakness, seizures, conductive hearing loss, and cardiac defects and irregularities of the skin, hair, and nails. It is primarily an autosomal dominant mutation in the human gap junction alpha 1 (GJA1) gene encoding for connexin 43, which has a high degree of penetrance but with considerable variability even within families [1,8].
Our patient had ODDD features including long thin nose, micropthalmia, microcorneas, microdontia, syndactyly, camptodactyly, and developmental delays. There are many syndromes with many similar phenotypes but this does not fit any one syndrome exactly. It is like ODDD but not exactly, and with no Ch. 6 abnormality seen. It is also similar to CFC syndrome which has been postulated to have deletions in the same region, however does not exactly fit the CFC syndrome. This is the first report of a patient with phenotypic features similar to those of ODDD syndrome with a microdeletion on chromosome 12q21, expanding the phenotype of ODD previously reported.
There have been 11 previous reports of patients with a deletion in or around this 12q21 region all of which have varying presentations but do contain some phenotypic similarities. Watson et al. presented a patient in 1989 with a deleted region of 12q15q21 who was not classified with any specific syndrome [5]. The patient had non-specific finding, but did have a long thin nose, syndactyly, and developmental delays. Rauen et al. report in 2000 of a patient with a deletion in 12q21.2q22 was more typical of CFC syndrome and included a short nose, cardiac defects, and fine, sparse hair and facial hair [2]. Rauen et al. 2002 report was non-specific but did share some features with their 2000 report including the deleted region 12q21.2q22, short nose, and sparse hair. However, unlike CFC syndrome this patient also had syndactyly and developmental delays. This patient was also born with a head circumference in the 10th percentile but later grew into normal ranges [3]. James et al. in 2005 presented a patient with a deletion in 12q21.2q22 with non-specific findings of low birth weight in the 3rd percentile, microcephaly, developmental and speech delays, strabismus, astigmatism, and narrow hands [4]. Even though these finding were non-specific they can be classified within the spectrum of an ODDD-like phenotype. Common findings in all of these reports were a hyperkeratotic rash, low set ears, and high arched palate. Table 1 lists some common features seen in ODD or CFC.
Table 1. List of some common features seen in ODD or CFC
Major Clinical Features |
OculoDentoDigital Dysplasia (ODDD) |
CardioFacioCutaneous (CFC) |
Patient’s presentation |
Camptodactyly |
Seen in 90% of patients with |
Not seen or very uncommon |
Patient has camptodactyly of fifth finger and in toes. |
|
Seen in 90% of patients with ODD |
Not seen or very uncommon |
Partial syndactaly of 2nd and 3rd toes |
Abnormal white matter on MRI |
May be seen in some patients, but data in |
May have abnormalities, such as structural changes |
Patient had no abnormal white matter seen on MRI |
Carious teeth |
Seen in 90% of patients with ODD |
Not seen or very uncommon |
Patient had enamel hypoplasia and multiple dental carries. |
|
Seen in 90% of patients with ODD |
Not seen or very uncommon |
Patient has microcorneas and small eye globes seen on MRI |
Cognitive impairment |
Seen in 50% of patients with ODD |
Seen in 90% of patients with CFC |
Patient presented with developmental delays and later in life required special education classes |
2021 Copyright OAT. All rights reserv
Of the more than 20 genes contained in the deleted region, there are several genes responsible for the development of cilia including BBS10 and CEP290 genes, and protein components of the Extracellular Matrix and collagen proteins (Figure 7). This can be part of the reason that there have been 11 such deletions in this region with varying phenotypes but with similarities in ectodermal findings. This is a large chromosome area and we cannot be sure the exact location or cause cannot be confirmed at this time. More case reports are required and with the improved use of microarray analysis more cases should be forthcoming.
Patients who may have an ODDD-like phenotype but are negative for connexin-43 mutation should be evaluated for a 12q21 deletion. This report expands the molecular cause for ODDD and CFC, even opening the possibility that an ODDD syndrome with varying presentations and features is present with this chromosomal region containing the genes responsible for the phenotype.
References
- Jones KL (2006) Smith’s Recognizable Patterns of Human Malformation, (6thedn), Philadelphia: Elsevier inc. 131-302.
- Rauen KA, Albertson DG, Pinkel D, Cotter PD (2000) Cardio-facio-cutaneous syndrome phenotype in an individual with an interstitial deletion of 12q: identification of a candidate region for CFC syndrome. Am J Med Genet 93: 219-22. [Crossref]
- Rauen KA, Albertson DG, Pinkel D, Cotter PD (2002) Additional patient with del(12)(q21.2q22): further evidence for a candidate region for cardio-facio-cutaneous syndrome?. Am J Med Genet 110: 51-6. [Crossref]
- James PA, Oei P, Ng D, Kannu P, Aftimos S (2005) Another case of interstitial del(12) involving the proposed cardio-facio-cutaneous candidate region. Am J Med Genet A 136: 12-6. [Crossref]
- Watson MS, Mcallister-Barton L, Mahoney MJ, Breg WR (1989) Deletion (12)(ql5q21.2). J Med Genet 26: 343-345.
- Bardakjian T, Weiss A, Schneider A (2014) “Anophthalmia/Microphthalmia Overview;” 2004 Jan 29 [Updated 2006 May 26]. In: Pagon RA, Adam MP, Bird TD, et al., (Eds.), GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014.
- Van der K, Marjo S, Valk J (2005) Magnetic Resonance of Myleniation and Myelin Disorders, (3rdedn), New York: Springer Berlin Heidelberg.
- Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, et al. (2009) GJA1 Mutations, Variants, and Connexin 43 Dysfunction as it Relates to the Oculodentodigital Dysplasia Phenotype. Hum Mutat 30: 724–733. [Crossref]
- Oculodentodigital dysplasia | Disease | Symptoms | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program [Internet]. Rarediseases.info.nih.gov. 2016 [cited 20 March 2016]. Available from: https://rarediseases.info.nih.gov/gard/7239/oculodentodigital-dysplasia/resources/9#explanation.
- Oculodentodigital dysplasia | Disease | Symptoms | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program [Internet]. Rarediseases.info.nih.gov. 2016 [cited 20 March 2016]. Available from: https://rarediseases.info.nih.gov/gard/7239/oculodentodigital-dysplasia/resources/9#explanation.
- Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, et al. (2010) Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 133: 2971-2982. [Crossref]
- Pierpont ME, Magoulas PL, Adi S, Kavamura MI, Neri G, et al. (2014) Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 134: e1149-1162. [Crossref]