Viral hemorrhagic fevers are rare but the life-threatening hemorrhagic disorder associated with viral sepsis. The demise of the patient occurs due to severe inflammation, multi-organ dysfunction syndrome and hemorrhage associated with poorly-defined coagulopathy. Sepsis of several families of RNA viruses causes endothelial injury that orchestrates inflammation and multi-organ dysfunction including the liver. To address additional clinical and hematological features, a novel pathogenesis based on “two-activation theory of the endothelium” is proposed. Endothelial injury activates endothelial cells that promote various clinical syndromes such as consumptive thrombocytopenia, multi-organ dysfunction and thrombotic microangiopathy. Endotheliopathy initiates two independent molecular events at endothelial cells: 1) release of inflammatory cytokines and 2) activation of the platelet and exocytosis of unusually large von Willebrand factor multimers. The former triggers activation of inflammatory pathway and the latter mediates activation of microthrombotic pathway. In viral sepsis, the activation of inflammatory pathway causes inflammation, but the activation of microthrombotic pathway manifests as disseminated intravascular microthrombosis (DIT). The pathogenesis of viral hemorrhagic fevers is hepatic coagulopathy triggered by acute hepatic necrosis as a result of endotheliopathy-associated DIT, which also could manifest as TTP-like syndrome.
viral hemorrhagic fevers; disseminated intravascular coagulation (DIC); endotheliopathy; thrombocytopenia; multi-organ dysfunction syndrome (MODS); thrombotic thrombocytopenic purpura (TTP); TTP-like syndrome
New Terminology
Microthrombogenesis; Disseminated intravascular microthrombosis (DIT); Vascular microthrombotic disease (VMTD)
Viral hemorrhagic fevers commonly occur in several families of RNA viruses: Arenaviridae (e.g., Lassa), Bunyaviridae (e.g., Hanta), Filoviridae (e.g., Ebola and Marburg) and Flaviviridae (e.g., yellow fever and dengue). It is a life-threatening hemorrhagic disorder, but the pathogenesis of hemorrhagic disorder is poorly understood [1].
Clinical features of viral hemorrhagic fevers include inflammatory symptoms such as fever, myalgia, arthralgia, malaise and weakness. Hemorrhagic signs are petechiae, bleeding in internal organs and external bleeding from bodily orifices like the mouth, eyes, or ears. Some patients develop bloody diarrhea. Eventually critically ill patients could progress to more serious conditions including seizures, delirium, shock, renal failure, acute respiratory distress and multi-organ dysfunction.
Potential causes of the hemorrhagic disease include: 1) thrombocytopenia related to bone marrow suppression or platelet destruction secondary to endothelial activation from viral sepsis [2,3], 2) disseminated intravascular coagulation (DIC) [1,3-5], and 3) hepatic coagulopathy associated with virus-induced hepatitis/hepatic necrosis [1,6,7], However, no credible clinical and laboratory data have been documented to explain the underlying coagulopathy.
Although viral hemorrhagic fevers commonly occur with etiology-undetermined thrombocytopenia in critically ill patients (TCIP) [1-3,6-9], its relationship to bleeding is not clearly determined because thrombocytopenia is typically mild to moderately severe and it alone can’t be accountable for the severe hemorrhagic disorder. Thus the thrombocytopenia has not entered as a serious issue in caring of viral hemorrhagic fevers other than platelet transfusion to maintain it at a safe level.
It is well known that the critical illnesses due to pathogens from bacterial, viral, fungal or parasitic sepsis are oftentimes associated with TCIP [10]. This term has been applied to etiology-undetermined thrombocytopenia after exclusion of known causes of acute thrombocytopenia (e.g., heparin-induced, drug or transfusion-associated, DIC-associated, hypersplenism-related, etc.). An interesting finding is TCIP not only occurs in sepsis/septic shock, but also occurs in other critical illnesses (e.g., severe trauma, complications of surgery, pregnancy and transplant, and immunologic and collagen vascular diseases) [10-13]. Recently, significant correlation was noted between the degree of thrombocytopenia and severity of the disease, and TCIP influenced the prognosis and likelihood of recovery [14,15]. Severer thrombocytopenia has been associated with systemic inflammatory response syndrome (SIRS) and multi-organ dysfunction syndrome (MODS) [16,17]. These observations support TCIP is an important participant in the pathogenesis of the critical illness including viral hemorrhagic fevers.
Viral hemorrhagic fevers are known to cause the injury to endothelial cells (ECs) leading to endotheliopathy and endothelial dysfunction, and endotheliopathy triggers multiple molecular events [18-20]. According to novel thesis of the “two-activation theory of the endothelium” (Figure 1) [6], endotheliopathy promotes the activation of two independent endothelial pathways (i.e., inflammatory and microthrombotic). In short, two important molecular events are: 1) release of inflammatory cytokines (e.g., interleukin (IL)-1, IL-6, tumor necrosis factor-a, and others) [20-22], and 2) activation of the platelet and exocytosis of unusually large von Willebrand factor multimers (ULVWF) [23-25]. The former triggers inflammation through “activation of inflammatory pathway”, and the latter mediates microthrombogenesis via “activation of microthrombotic pathway” as illustrated in Figure 1. In endotheliopathy, microthrombogenesis is the process in which long elongated ULVWF strings are anchored to ECs to recruit activated platelets, and to assemble and decorate platelet-ULVWF complexes as microthrombi [25-27]. This results in disseminated intravascular microthrombosis (DIT) triggering thrombotic thrombocytopenis purpura (TTP)-like syndrome.
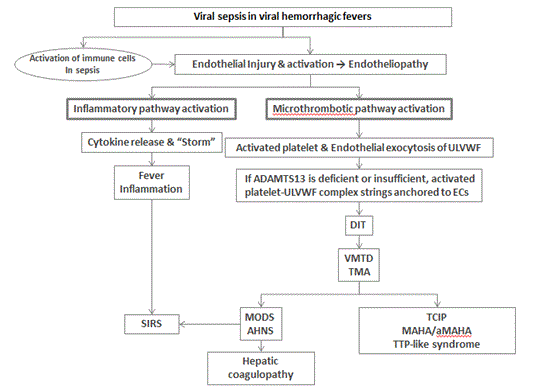
Figure 1. Pathogenesis of endotheliopathy-associated DIT/TTP-like syndrome in viral hemorrhagic fevers.
AHNS, acute hepatic necrosis syndrome; aMAHA/MAHA, atypical microangiopathic hemolytic anemia/microangiopathic hemolytic anemia; ARDS, acute respiratory distress syndrome; DIT, disseminated intravascular microthrombosis; ECs, endothelial cells; MODS: multi-organ dysfunction syndrome; MTA, microthrombotic angiopathy; SIRS, systemic inflammatory response syndrome; TTP, thrombotic thrombocytopenic purpura; VMTD, vascular microthrombotic disease; ULVWF, unusually large von Willebrand factor multimers.
DIT is the underlying pathological condition leading to vascular microthrombootic disease (VMTD). Systemic VMTD includes two clinical disorders: thrombotic thrombocytopenic purpura (TTP) and TTP-like syndrome. In TTP, microthrombogenesis occurs in circulation due to hyperactivity of ULVWF in both hereditary and antibody-associated type, but in TTP-like syndrome developing in viral hemorrhagic fevers and other critical illnesses, it occurs at the intravascular surface of ECs. The different pathogenesis and clinical characteristics of TTP and TTP-like syndrome are summarized in Table 1. In the critical illness DIT is made of microthrombi that consist of platelet-ULVWF complexes and is anchored to ECs. DIT as a result of endotheliopathy in viral hemorrhagic fevers can be called endotheliopathy-associated DIT/VMTD.
Table 1. Acquired vascular microthrombotic disease (DIT/VMTD): pathogenic and clinical characteristics of TTP and TTP-like syndrome.
|
Antibody-associated DIT/VMTD
(Acquired TTP) |
Endotheliopathy-associated DIT/VMTD
(TTP-like syndrome) |
Etiology
Probable pathogenesis
|
Anti-ADAMTS13 antibody
¯
Increased destruction of ADAMTS13
¯
Hyperactive mULVWF ® Aggregate with platelet
¯
Microthrombosis |
ECs injury leading to their dysfunction due to sepsis, surgery, trauma, Shiga toxin, preeclampsia, cancer, drugs, and others
¯
Release of eULVWF and anchored to ECs
¯
Decorated eULVWF with platelets
¯
Microthrombosis |
ADAMTS13 level |
Usually <5% |
Usually 20-70% |
ADAMTS13 antibody |
Positive |
Negative |
Intravascular ULVWF |
Increased |
Increased |
Thrombocytopenia |
Present |
Present |
Anemia |
MAHA |
MAHA/aMAHA |
Schistocytes |
Always present at usually >2% |
Typically be present with fewer schistocytes |
Hypoxic organ dysfunction |
Present |
Present |
Typical examples of involving organs
|
Brain (CNSD); Kidneys (ARF)
|
Brain (CNSD); lungs (ARDS); liver (HELLP);
kidneys (HUS); bowels (GIHS); liver (AHNS);
adrenals (Waterhouse-Friderichsen syndrome)
muscle (rhabdomyolysis); skin (purpura fulminans) |
Advanced stage of organ involvement |
MODS |
SIRS; MODS |
Coagulopathy |
Absent |
Absent unless hepatic coagulopathy occurs |
Response to TPE |
Very good response |
Excellent response if treated early |
Platelet transfusion |
Contraindicated |
Contraindicated |
DIT, disseminated intravascular microthrombosis; VMTD, vascular microtrombotic disease; TTP, thrombotic thrombocytopenic purpura; SIRS, systemic inflammatory response syndrome; CNSD, central nervous system dysfunction; ARDS, acute respiratory distress syndrome; GIHS, gastrointestinal hemorrhagic syndrome; AHNS, acute fulminant hepatitis/acute hepatic necrosis syndrome; ARF, acute renal failure; HUS, hemolytic uremic syndrome; TPE, therapeutic plasma exchange; mULVWF, megakaryocytic unusually large von Willebrand factor multimers; eULVWF, endothelial ULVWF; TMA, thrombotic microangiopathy; MAHA, microangiopathic hemolytic anemia; aMAHA, atypical microangiopathic hemolytic anemia; ECs, endothelial cells
In viral hemorrhagic fevers, endotheliopathy-associated DIT/VMTD could trigger TTP-like syndrome [6,28-31], which is characterized by consumptive thrombocytopenia, microangiopathic hemolytic anemia (MAHA)/atypical MAHA (aMAHA) (if schistocytes are fewer) and hypoxic organ dysfunction syndromes. Unlike true DIC in which hemostatic (coagulation) disorder occurs following tissue factor (TF) pathway activation, endotheliopathy-associated DIT/VMTD is the microthrombotic disorder occurring as a result of microthrombogenesis. In endotheliopathy-associated DIT/VMTD, TF pathway activation is not involved and thus coagulation factors are not consumed and depleted.
The simple answer is no. Viral hemorrhagic fevers have been attributed to “DIC” [3-5,32], mainly utilizing the International Society on Thrombosis and Haemostasis (ISTH) DIC-scoring system and accepting the microthrombosis in the critically ill patient as the marker for hemostatic (coagulation) disorder. This diagnosis hasn’t been based on more reliable coagulation factor assay of FVIII and FV, which are typically depleted in true DIC as seen in acute promyelocytic leukemia [33].
Donald McKay in early1950s coined the term “DIC” [34] for a microthrombootic disorder, which he interpreted as a coagulation disorder. He and his followers have believed intravascular hyaline microthrombi in the luminal arterioles and capillaries in the pathologic tissue examination consist of micro-clots of platelets, coagulation factors and fibrins. In coagulation profile, the supporting evidence is prolonged prothrombin time, prolonged activated partial thromboplastin time, hypofibrinogenemia, and increased fibrin degradation products. In many patients with “DIC”, however, coagulation profile has been perfectly normal and hemorrhagic tendency does not occur. Puzzled but conveniently, the concept of “chronic/compensated/covert” was introduced. This description, however, cannot explain inexplicably extensive microthrombi in the absence of depleted coagulation factors.
Clinically “DIC” and endotheliopathy-associated DIT/VMTD (i.e., TTP-like syndrome) are the same in their underlying risk factors and presentation. Both “DIC” and TTP-like syndrome almost always occur in critical illnesses (e.g., sepsis/septic shock, trauma, immunologic and collagen-vascular diseases, and complications of surgery, pregnancy and transplant) [35, 36]. In both disorders, the pathology is characterized by arteriolar and capillary hyaline microthrombi with variable fibroblastic proliferation [37, 38]. Hematological features are also the same with consumptive thrombocytopenia and MAHA/aMAHA. Thus “DIC” and endotheliopathy-associated DIT/VMTD must be the same disorder.
According to the “two-activation theory of the endothelium”, DIT induced by microthrombogenesis is completely different from true DIC occurring as a result of activated TF coagulation pathway. Their characteristic differences are shown in Table 2. The former is a microthrombotic disorder, but the latter is a hemostatic (coagulation) disorder. Considering the difference between “DIC” and DIT in their currently accepted pathogenic mechanisms, “DIC” must have been understood with a wrong concept. Hence, “DIC” is a misnomer. For more than 60 years, this unfortunate misconception of “DIC” has created confusion in medical science and practice, including diagnostic dilemma and treatment failures until this day [39-41].
Table 2. Hematological and clinical characteristics of endotheliopathy-associated DIT/VMTD and true DIC.
|
Endotheliopathy-associated DIT
(including “DIC” of McKay) |
True DIC |
Examples |
TTP-like syndrome |
DIC associated with APL |
Nature of the disorder |
Microthrombosis made of platelet-ULVWF complexes |
Coagulation activated by TF-FVIIa complexes |
Mechanism of the genesis |
Intravascular microthrombogenesis |
Intravascular coagulation |
Inciting events |
Sepsis, complications of surgery, pregnancy, cancer,
and transplant, and drugs/toxins leading to endotheliopathy |
APL and drugs (?) leading to TF expression |
Hematological manifestations |
TTP-like syndrome |
Hemorrhagic disorder of APL |
Pathogenesis
Mechanism
Site of activation
Pathology
Result of pathogenesis |
Activation of microthrombotic pathway
Intravascular surface of the endothelium
Endothelial activation/dysfunction ® endotheliopathy
Formation of platelet-ULVWF microthrombi |
Activation of TF-FVIIa complex coagulation pathway
In circulation of the Intravascular space
TF expression ® coagulation and factor consumption Depletion of fibrinogen, FVIII, FV |
Essence of pathology |
Arteriolar and capillary luminal hyaline microthrombi |
Incoagulable blood/unstable blood clots |
Effect on the involved organs |
Vascular microthrombosis leading to organ hypoxia |
Hemorrhage leading to organ damage |
Coagulation tests
Fibrinogen; PT; aPTT; TT
FDP
FVIII activity
Thrombocytopenia |
Normal
Normal
Normal or increased
Moderately severe |
Prolonged
Increased
Markedly decreased
Mild to very severe |
Associated clinical syndromes |
TTP-like syndrome
AHNS with hepatic coagulopathy
MODS
SIRS |
Hemorrhagic disorder |
Associated hematologic features
Schistocytes
MAHA/aMAHA
Consumptive thrombocytopenia
Hepatic coagulopathy |
0 - +++
Often present
Always present
May occur |
0 - + (?)
Absent
Present (?)
Unusual |
Incidence in clinical practice |
Very common |
Extremely rare |
Therapy
Platelet transfusion
Treatment |
Contraindicated
TPE; rADAMTS13 (expected to be very effective) |
May be needed for APL
Treat underlying pathology (e.g., ATRA in APL) |
APL, acute promyelocytic leukemia; aPTT, activated partial thromboplastin time; aMAHA/MAHA, atypical microangiopathic hemolytic anemia/MAHA; ATRA, All-trans retinoic acid; DIC, disseminated intravascular coagulation; DIT, disseminated intravascular microthrombosis; ULVWF, endothelial unusually large von Willebrand factor multimers; FV, factor V; FVIIa, activated factor VII; FVIII, factor VIII; FDP, fibrin degradation products; MTA, microthrombotic angiopathy; PT, prothrombin time; TF, tissue factor; TPE, therapeutic plasma exchange; TT, thrombin time; MODS, multi-organ dysfunction syndrome; rADAMTS13, recombinant ADAMTS13; SIRS, systemic inflammatory response syndrome; VMTD, vascular microthrombotic disease
It should be emphasized that no single laboratory test or set of tests is sensitive or specific enough to allow a definite diagnosis of “DIC” [39]. In most cases the diagnosis is based on the combination of results of non-specific laboratory test results in the patient with a clinical condition known to be associated with “DIC” [41].
If one understands and accepts the fact that “DIC” is a misnomer but one accepts it as endotheliopathy-associated DIT/VMTD, viral hemorrhagic fevers can be explained perfectly well by the concept of DIT. The next question is how viral hemorrhagic fevers get the hemorrhagic disorder. Another word, What is the correct diagnosis for “DIC” that is associated with abnormal coagulation profile? In viral hemorrhagic fevers, acute fulminating hepatitis/acute hepatic necrosis, especially multifocal necrosis type, occurs regularly without a good explanation [42-46]. Acute fulminant hepatitis and hepatic necrosis are not due to viral hepatitis, but are due to DIT. The “two-activation theory” supports hepatic coagulopathy could occur due to endotheliopathy-associated DIT/VMTD causing multiple hepatic microinfarctions (AHNS) as noted in Figure 1 [6]. Indeed, hepatic coagulopathy shows exactly the same coagulation profile as seen in “acute DIC”.
True DIC is very rare but occurs perhaps in acute promyelocytic leukemia, presumably due to TF expression from leukemic cells [47]. As illustrated in Table 2, the predominant feature of true DIC is hemorrhage without MAHA/aMAHA and hypoxic organ dysfunction [33,47,48]. This supports that MAHA/aMAHA, MODS and TTP-like syndrome are the manifestations of endotheliopathy-associated DIT.
In differentiating true DIC from hepatic coagulopathy, the most important test is the assay of coagulation factors: especially for depleted FVIII and FV in true DIC, increased FVIII and markedly decreased liver dependent FVII in hepatic coagulopathy. A suggested guideline for laboratory tests is presented in Table 3 to aid in differential diagnosis among complicated thrombopathies and coagulopathies [6].
Table 3. Differential characteristic hematologic features among thrombopathies and coagulopathies (Adapted from Chang JC (6) with permission).
|
TTP & TTP-like syndrome (DIT) |
TTP-like syndrome (DIT) associated
with HC (e.g., Ebola) = acute “DIC” of McKay |
DIC (e.g., acute promyelocytic leukemia) |
PF (e.g., amyloidosis) |
Thrombocytopenia |
Always present |
Always present |
Always present |
Not present |
MAHA/aMAHA |
Almost always present |
Usually present |
Very unlikely to be present |
Not present |
Fibrinogen |
Normal |
Decreased |
Always decreased |
Always decreased |
Factor VIII |
Normal |
Normal or increased |
Markedly decreased |
Decreased |
Factor V |
Normal |
Decreased |
Decreased |
Decreased |
Factor X |
Normal |
Decreased |
Usually normal |
Normal |
Factor VII |
Normal |
Markedly decreased |
Normal |
Normal |
Factor IX |
Normal |
Decreased |
Normal |
Normal |
FDP |
Normal |
Positive |
Positive |
Strongly positive |
Thrombin time |
Normal |
Prolonged |
Prolonged |
Prolonged |
Thrombosis form |
Microthrombi |
Microthrombi |
Friable macrothrombi (?) or not formed |
Absent |
Bleeding: Character |
Rare, mild petechiae |
May cause serious bleeding |
Common, serious bleeding |
Slow & persistent bleeding |
Treatment |
Usually no need of treatment |
Controllable with FFP |
Abrogated with ATRA & chemotherapy |
Treatable with AFA |
Platelet transfusion |
Contraindicated |
Contraindicated |
May be used with ATRA |
Not needed |
TTP, thrombotic thrombocytopenic purpura; HC, hepatic coagulopathy; DIT, disseminated intravascular microthrombosis; DIC, disseminated intravascular coagulation; PF, primary fibrinolysis; MAHA, microangiopathic hemolytic anemia; aMAHA, atypical MAHA; FFP, fresh frozen plasma; AFA, anti-fibrinolytic agents; ATRA, all-trans retinoic acid
In viral hemorrhagic fevers, TCIP is the earliest indicator suggesting that microthrombogenesis is in progress. If the hemorrhagic disorder occurs, it is not due to true DIC nor is likely due to thrombocytopenia, but most likely is due to hepatic coagulopathy. The “two activation theory” not only explains concomitant inflammation, TCIP and progressive hypoxic organ dysfunction, but also would help to unmask unrecognized syndromes such as impending cytokine “storm”, TTP-like syndrome, MAHA/aMAHA, MODS and SIRS in viral hemorrhagic fevers.
Viral hemorrhagic fevers are not due to “DIC” but are due to endotheliopathy-associated DIT/VMTD, which hematologic manifestation could lead to TTP-like syndrome. The treatments for viral hemorrhagic fevers are fresh frozen plasma for hepatic coagulopathy and therapeutic plasma exchange for TTP-like syndrome if the diagnosis is confirmed [6]. If therapeutic plasma exchange is not available, clinical trials using anti-microthrombotic agents such as recombinant ADAMTS13 and N-acetyl cysteine may be considered. Platelet transfusion and anticoagulation therapy are contraindicated.
None
The author Jae C. Chang, M.D. has neither actual nor potential personal or financial conflicts of interest in regard to this article.
- Paessler S, Walker DH (2013) Pathogenesis of the viral hemorrhagic fevers. Annu Rev Pathol 8: 411-440. [Crossref]
- Perng GC (2012) Role of Bone Marrow in Pathogenesis of Viral Infections. J Bone Marrow Res 1. [Crossref]
- Zapata JC, Cox D, Salvato MS (2014) The role of platelets in the pathogenesis of viral hemorrhagic fevers. PLoS Negl Trop Dis 8: e2858. [Crossref]
- Sundberg E, Hultdin J, Nilsson S, Ahlm C (2011) Evidence of disseminated intravascular coagulation in a hemorrhagic fever with renal syndrome-scoring models and severe illness. PLoS One 6: e21134.
- Geisbert TW, Young HA, Jahrling PB (2003) Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol 163: 2371-2382.
- Chang JC (2016) A Thought on Possible Pathogenesis of Ebola Viral Hemorrhagic Disease and Potential Treatments: Could it be Thrombotic Thrombocytopenic Purpura-like Syndrome? J Ther Aph Dialysis 20: 93-98.
- CDC: Ebola Virus Disease (EVD) Information for Clinicians in U.S. Healthcare Settings.
- Rashmi MV, Hamsaveena (2015) Haematological and biochemical markers as predictors of dengue infection. Malays J Pathol 37: 247-251.
- Hu JL, Li ZF, Wang XC (2016) Risk Factors for Bunyavirus-Associated Severe Fever with Thrombocytopenia Syndrome: A Community-Based Case-Control Study. PLoS One 11: e0166611.
- Williamson DR, Albert M, Heels-Ansdell D (2013) PROTECT collaborators.; Canadian Critical Care Trials Group.; Australian and New Zealand Intensive Care Society Clinical Trials Group. Thrombocytopenia in critically ill patients receiving thromboprophylaxis: frequency, risk factors, and outcomes. Chest 144: 1207-1215.
- Carrick MM, Tyroch AH, Youens CA, Handley T (2005) Subsequent development of thrombocytopenia and coagulopathy in moderate and severe head injury: support for serial laboratory examination. J Trauma 58: 725-729.
- Chen Z, Liang MY, Wang JL (2011) [Etiology and clinical characteristics of pregnancy-emerged thrombocytopenia]. Zhonghua Fu Chan Ke Za Zhi 46: 834-839. [Crossref]
- Naqvi TA, Baumann MA, Chang JC (2004) Post-operative thrombotic thrombocytopenic purpura: a review. Int J Clin Pract 58: 169-172.
- Levi M (2016) Platelets in Critical Illness. Semin Thromb Hemost 42: 252-257. [Crossref]
- Venkata C, Kashyap R, Farmer JC, Afessa B (2013) Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. J Intensive Care 1: 9.
- Ogura H, Gando S, Iba T (2007) Japanese Association for Acute Medicine Disseminated Intravascular Coagulation Study Group. SIRS-associated coagulopathy and organ dysfunction in critically ill patients with thrombocytopenia. Shock 28: 411-417.
- Stravitz RT, Ellerbe C, Durkalski V, Reuben A, Lisman T, et al. (2016) Acute Liver Failure Study Group. Thrombocytopenia Is Associated With Multi-organ System Failure in Patients With Acute Liver Failure. Clin Gastroenterol Hepatol 14: 613-620.e4.
- Peters CJ, Zaki SR (2002) Role of the endothelium in viral hemorrhagic fevers. Crit Care Med 30: S268-273. [Crossref]
- Bodur H, Akinci E, Ongürü P (2010) Evidence of vascular endothelial damage in Crimean-Congo hemorrhagic fever. Int J Infect Dis 14: e704-7.
- Srikiatkhachorn A, Spiropoulou CF (2014) Vascular events in viral hemorrhagic fevers: a comparative study of dengue and hantaviruses. Cell Tissue Res 355: 621-633. [Crossref]
- Aird WC (2003) The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101: 3765-3777. [Crossref]
- Xing K, Murthy S, Liles WC, Singh JM (2012) Clinical utility of biomarkers of endothelial activation in sepsis--a systematic review. Crit Care 16: R7.
- Janicek MJ, Van den Abbeele AD, Hollenberg NK, Kassis AI, Holman BL, et al. (1990) Platelet activation and aggregation after endothelial injury. Assessment with indium-111-labeled platelets and angiography. Invest Radiol 25: 988-993.
- Bockmeyer CL, Claus RA, Budde U, Kentouche K, Schneppenheim R, et al. (2008) Inflammation-associated ADAMTS13 deficiency promotes formation of ultra-large von Willebrand factor. Haematologica 93: 137-140. [Crossref]
- Valentijn KM, van Driel LF, Mourik MJ, Hendriks GJ, Arends TJ, et al. (2010) Multigranular exocytosis of Weibel-Palade bodies in vascular endothelial cells. Blood 116: 1807-1816. [Crossref]
- De Ceunynck K, De Meyer SF, Vanhoorelbeke K (2013) Unwinding the von Willebrand factor strings puzzle. Blood 121: 270-277. [Crossref]
2021 Copyright OAT. All rights reserv
- Padilla A, Moake JL, Bernardo A (2004) P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood 103: 2150-2156.
- Vaziri S, Navabi J, Afsharian M (2008) Crimean congo hemorrhagic fever infection simulating thrombotic thrombocytopenic purpura. Indian J Hematol Blood Transfus 24: 35-38.
- Deepanjali S, Naik RR, Mailankody S, Kalaimani S, Kadhiravan T (2015) Dengue Virus Infection Triggering Thrombotic Thrombocytopenic Purpura in Pregnancy. Am J Trop Med Hyg 93: 1028-1030. [Crossref]
- Ardalan MR, Tubbs RS, Chinikar S, Shoja MM (2006) Crimean-Congo haemorrhagic fever presenting as thrombotic microangiopathy and acute renal failure. Nephrol Dial Transplant 21: 2304-2307.
- Lopes da Silva R (2011) Viral-associated thrombotic microangiopathies. Hematol Oncol Stem Cell Ther 4: 51-59.
- Peters CJ, Liu CT, Anderson GW Jr, Morrill JC, Jahrling PB (1989) Pathogenesis of viral hemorrhagic fevers: Rift Valley fever and Lassa fever contrasted. Rev Infect Dis 4: S743-S749.
- Cooperberg AA (1967) Acute promyelocytic leukemia. Can Med Assoc J 97: 57-63. [Crossref]
- ISTH International Society on Thrombosis and Haemostasis [Cited from: http://slideplayer.com/slide/3080693/]
- Franchini M, Lippi G, Manzato F (2006) Recent acquisitions in the pathophysiology, diagnosis and treatment of disseminated intravascular coagulation. Thromb J 4: 4. [Crossref]
- Nguyen TC, Kiss JE, Goldman JR, Carcillo JA (2012) The role of plasmapheresis in critical illness. Crit Care Clin 28: 453-468.
- Sueishi K, Takeuchi M (1993) [Pathology of disseminated intravascular coagulation]. Nihon Rinsho 51: 30-36. [Crossref]
- Tsai HM (2010) Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol 91: 1-19. [Crossref]
- Slofstra SH, Spek CA, ten Cate H (2003) Disseminated intravascular coagulation. Hematol J 4: 295-302. [Crossref]
- Gando S (2012) The utility of a diagnostic scoring system for disseminated intravascular coagulation. Crit Care Clin 28: 373-388. [Crossref]
- Franchini M, Lippi G, Manzato F (2006) Recent acquisitions in the pathophysiology, diagnosis and treatment of disseminated intravascular coagulation. Thromb J 4: 4.
- Kularatne SA, Imbulpitiya IV, Abeysekera RA, Waduge RN, Rajapakse RP, et al. (2014) Extensive haemorrhagic necrosis of liver is an unpredictable fatal complication in dengue infection: a postmortem study. BMC Infect Dis 14: 141.
- Talwani R, Gilliam BL, Howell C (2011) Infectious diseases and the liver. Clin Liver Dis 15: 111-130.
- Samanta J, Sharma V (2015) Dengue and its effects on liver. World J Clin Cases 3: 125-131. [Crossref]
- El Sayed SM, Abdelrahman AA, Ozbak HA, Hemeg HA, Kheyami AM, et al. (2016) Updates in diagnosis and management of Ebola hemorrhagic fever. J Res Med Sci 21: 84. [Crossref]
- Sharingbutton (2014). Global Ebola-Are We Prepared? Emergency Medicine. 46: 440-449.
- Tallman MS, Hakimian D, Kwaan HC, Rickles FR (1993) New insights into the pathogenesis of coagulation dysfunction in acute promyelocytic leukemia. Leuk Lymphoma 11: 27-36. [Crossref]
- Chang JC, Gross HM, Jang NS (1990) Disseminated intravascular coagulation due to intravenous administration of hetastarch. Am J Med Sci 300: 301-303. [Crossref]