Nowadays, understanding complex association among HLA-B27, ER stress pathways and Spondyloarthropathies (SpAs) has been considered to be important situation. Hitherto, many theories have been claimed by researchers in order to explain this association, but HLA-B27 misfolding theory can be the key theory among all theories. HLA-B27 misfolding is related to UPR (Unfolded Protein Response) and EOR (ER Overload Mechanism), since UPR mechanism protect ER homeostasis against misfolded protein and EOR system provide normal ER function to work on accumulating protein. On the other hand, ER associated degradation (ERAD) degrades unfolded or misfolded proteins which are re-translocated to cytosol for degradation. Present work aims to summarize the main pathways of UPR and ERAD, and then to reveal the relation between the tendency of HLA-B27 to misfold, ER stress and SpAs, especially Ankylosing Spondylitis (AS).
ER stress, HLA-B27 misfolding, spondyloarthropathies, ankylosing spondylitis
ER is a system of membranes, lies between cell membrane and nucleus in most eukaryotic cells. ER contains small vesicles called cisterns and tubules made of membranes. ER is named as rough ER or granulated ER and agranulated ER depending on containing ribosome on its outer surface or not, respectively. Because ER participates in important cellular events as protein folding, biosynthesis of lipids, intracellular membrane trafficking and intracellular calcium signaling; ER is in the center of many studies [1-3].
The process that interrupts the physiologic functions of ER caused by several factors as redox, pH change, hyperthermia, etc., is called ER stress. First of all, the misfolded proteins are exported back to cytoplasm for proteasomal degradation. The process is called ER-associated degradation (ERAD). If protein accumulation occurs via impairment in proper protein folding and forming unfolded and misfolded proteins, ER stress response is initiated. ER stress stimulates the Unfolded Protein Response (UPR) for recovering the stress conditions [4-6]. The UPR orchestrate is initiated by three transmembrane proteins which are localized in ER in physiologic conditions and inhibited by glucose related protein 78 (Grp78) binding: Inositol Requiring 1 (IRE1), Protein Kinase R-like ER Kinase (PERK), and Activating Transcription Factor 6 (ATF6) [7]. Figure 1 summarizes the UPR and ERAD pathways.
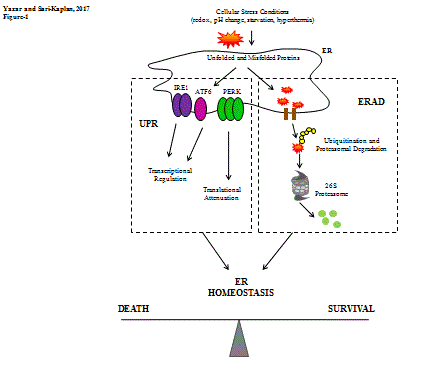
Figure 1. Unfolded and misfolded proteins are exported back to cytoplasm for degradation by proteasomal system; the process is called ERAD system. Also unfolded and misfolded protein accumulation cause UPR activation. UPR pathway is organized by three members: IRE1, PERK and ATF6.
The inhibitory molecule, Grp78, was first discovered in fibroblastic glucose-free cell culture medium [8]. Grp78 is also referred as immunoglobulin binding protein (BiP) [9]. Grp78 is one of the ER molecular chaperons that assist protein folding and localized in ER lumen [10-13]. It has 60% amino acid homology with Heat Shock Protein-70 (HSP70); homology domain also contains ATP-binding domain which is necessary for its chaperon activity [14]. Main activity of Grp78 in ER stress signaling is that it has inhibitory effect on UPR regulators IRE1, PERK and ATF6 during stress-free conditions. After having stress signals, Grp78 dissociates from UPR regulators and UPR regulators become at ‘on state’ [15]. Proper protein folding is achieved by N-linked protein glycosylation and dissociation of Grp78 is also related with N-glycosylation. N-glycosylation is a co-translational and also a post-translational modification of protein folding. Glucose or mannose binding via N-glycosylation generates a docking site for molecular chaperons, calreticulin and calnexin, to help proper protein folding. After forming intra-molecular disulfide bonds and non-covalent bonds, carbohydrate group is released by glycosidase-II enzyme and folded protein is sent to cytosol. If proper folding is not formed, protein is sent to folding process again or sent to ERAD pathway for ubiquitin- and ATP-dependent degradation by proteasomal system. Unfolded or misfolded proteins have chemo-attractive sites for Grp78 so that Grp78 can leave the ER-resident transmembrane UPR regulator molecules and attach to unfolded or misfolded proteins which cause UPR pathway activation in response [6,16-19].
ER and Golgi together with proteasomal system have crucial roles in immune system as sampling protein pool, antigen loading to human leukocyte antigen (HLA) molecules and antigen presentation on cell surface. So those ER and proteasomal system are important research areas in immune system diseases [20]. As an example, ankylosing spondylitis (AS), which is an autoinflammatory disease, has been shown to be related with ER stress and HLA processing. Instead of undefined molecular mechanism of AS and several suggested hypothesis, one of the most accepted theory is HLA-B27 misfolding and ER stress. In the present work, the current knowledge on the HLA-B27 folding, misfolding and their relation with AS pathogenesis will be summarized.
HLA-B27, encoded in HLA-B locus at located on the short (p) arm of chromosome 6 is a class of MHC class I molecule. Generally, peptide presentation, formed from intracellular antigens, to CD8+ cytotoxic T cells and acting as a ligand for Natural Killer cells are crucial functions of MHC class I molecules. MHC class I molecules, which are glycoproteins which can fold within the lumen of the endoplasmic environment, in ER, are comprised a heavy chain noncovalently associated with the light chain beta-2-microglobulin (β2m) and loaded with a 8–13 amino acid long peptide [21,22].
HLA Class I molecules especially HLA-B27 exhibit unique protein folding status that can conflict with ER quality control mechanism and have tendency to switch to misfolded state [23]. Other two unique features of HLA-B27, besides a tendency to misfolded state, such as peptide binding specificity and ability for forming heavy chain homodimers in cell surface recycling, three are also considered to diversify it from among all other MHC Class I molecules [24-25]. Tendency of HLA-B27 misfolding was discovered randomly with mutagenesis studies that claim B pocket in the peptide binding groove resulting in a slow folding phenotype and after ERAD of newly synthesized heavy chains in experiments with transfected EBV- transformed cells with a single copy of HLA-B27 [26].
HLA-B27 allele class is one of the most studied alleles among all HLA molecules since they generally have significant relation between Spondyloarthropathies [27]. Newly, importance of these molecules has enlarged with studies that protective role has been discovered in viral infection like AIDS and Hepatitis C [28,29]. HLA-B27 allele family has differences from each other with one or a few amino acids in their peptide binding groove region that have been reported to have an association [27].
Among all Spondyloarthropathies (SpAs) that are known as inflammatory arthritic conditions, HLA-B27 is associated to be presented most typically with Ankylosing Spondyltis (AS) that is an example of SpAs [30,31]. In all SpAs, MHC genes have been presented with 40-50% genetic tendency and especially approximately 30% of the tendency has been related with HLA-B27 [31-33]. Although, HLA-B27 expression ratio in AS patients is about 90-95%, the pathogenesis of AS with HLA-B27 association has not to be enlighten, yet [23]. The possible role of HLA-B27 in pathogenesis of SpAs has several difficulties to understand precisely, since genetic and environmental elements of autoimmune diseases presentation that are known as complex and multifactorial disorder, increase the heterogeneity [34]. Subsequently, many theories, such as arthritogenic peptide hypothesis, cell surface HLA-B27 homodimers hypothesis, enhanced survival of some microbes in HLA-B27 cells hypothesis have been suggested to enlighten the possible SpAs and HLA-B27 association but HLA-B27 misfolding is considered to be the most accepted theory. (Figure 2) Therefore, HLA-B27 misfolding theory is referred as the main focus of this paper.
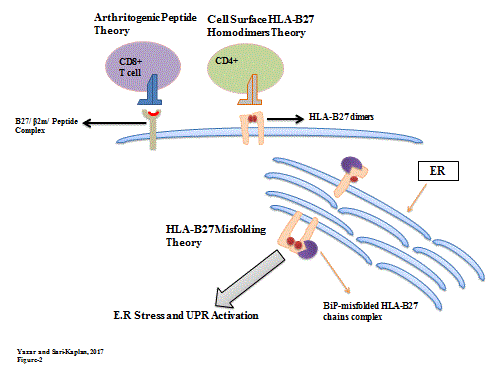
Figure 2. Three most important theories in pathogenesis of SpAs. i) Arithrogenic peptide theory: Properly fold forms of HLA-B27 can form complex with beta-2 microglobuline (β2m) and self-peptide selection and presentation; so it has been considered that this complex can be the target of autoreactive CD8+ T cells, resulting in inflammation. ii) Cell surface HLA-B27 homodimers theory: Randomly formed cell surface HLA-B27 dimers are hypothesized to be bind killer immunoglobulin receptors and trigger inflammation. iii) HLA-B27 misfolding theory: Misfolded HLA-B27 chains and after that BiP binding by newly synthesized HLA-B27 heavy chains causes ER stress, that is considered to be the key factor development of spondyloarthritis through UPR activation.
Up to now, biochemical studies indicate that HLA-B27 can show different biochemical pattern from most other MHC class I molecules. HLA-B27 molecules has been assumed that they have unique biochemical properties such as enhanced tendency to misfold and susceptible to aggregation [24,27]. As a comparison to other HLA-B molecules, the content of residues in B pocket, is the region where the molecule binds to N-terminal motif of MHC class I associated peptides, are quite remarkable and unique such as His9, Thr24, Glu45, Cys67, Lys70, Ala71, and Gln97. These residues are sorted in the B pocket and can bind the second anchor residue of associated peptides. Firstly, this phenomenon was considered an evidence for HLA-B27-specific arthritogenic peptides theory but artificially placed of these residues into other MHC class I molecules concluded in increased misfolding of non-SpA-associated HLA molecules [35].
Other key factor for understanding HLA-B27 misfolding with biochemical characteristics is formation of a result of slow heavy chain folding and aberrant disulfide bond formation and also after increased exposure of unpaired Cys residues in oxidized ER environment [25]. Besides enhanced tendency of misfolding, HLA-B27 was demonstrated that it can form aggregates in a study of purified recombinant protein [24]. Also, HLA-B27 dimers can be correlated with disease prevalence in the HLA-B27 transgenic rat models, so these aggregate structures may be responsible for “toxicity gain of function” [36].
Generally, HLA-B27 dimerization was considered to be formed at p67 from an unpaired cysteine which constitutes part of the peptide binding groove. This Cys67 residue is thought to be quite important in dimerization at the cell surface; while dimerization in the ER can occur through structurally conserved cysteine at p164 [37]. In a study with transgenic rats, mutant HLA-B27 molecule without Cys67 is demonstrated to still develop disease but in a lower incidence. Therefore, Cys67 is a crucial residue and plays important role in dimer formation of HLA-B27 and HLA-B27 dimers can be considered as a disease marker for SpAs [38].
The history of HLA-B27 misfolding hypothesis started in the mid-1990s with a crucial biological discovery. The observation of global protein machinery impairment in the ER, which can induce cellular responses resulting in activation of genes and protein production to recover ER homeostasis, provide first hints for protein misfolding mechanism [39,40]. The terms for these cellular responses are ER stress responses that there are two types such as UPR and ER Overload Response (EOR). Generally, main goal in UPR is to protect ER homeostasis against misfolded protein; while EOR system deals with accumulating protein in order to provide normal ER function. In HLA-B27 misfolding hypothesis, HLA-B27 misfolding occurs within ER environment and can cause toxicity function by activation of UPR and EOR; also this phenomenon leads to impairments in normal cellular function and activation of proinflammatory cytokine production related to SpAs development [41] (Figure 3).

Figure 3. Cytokine upregulation involvement in HLA-B27 misfolding-E.R stress mechanism resulting UPR activation and inflammation. After IFNs induce increase of MHC Class I expression, UPR activation can be occur within antigen-presenting cells of HLA-B27-expressing individuals when a critical threshold of misfolding is reached. UPR pathway trigger can be trigger cytokine activation and release affecting helper T cells, while EOR system maintains ER homeostasis within ER environment. Helper T cells can trigger IL-17 responsive cell via IL-17 release and this activate IL-17 responsive cells to produce TNFα, IL-1 and IL-6; especially IL-1 can cause inflammation.
Generally, SpAs are classified as autoinflammatory disease rather than autoimmune disease and proinflammatory cytokine production related with HLA-B27 misfolding and ER stress is an interesting fact for HLA-B27 misfolding and SpAs association [22]. Autoinflammatory disorders are manifested with inflammation without autoantibodies and antigen specific T cells, which are two important conditions that SpAs surprisingly have [42]. Unlike UPR as stated before in this review, EOR has not been enlighten yet; however it has been considered that accumulation of misfolded protein in the ER can cause the activation of transcription factor NF-κB. Therefore, activation of NF-κB can explain the association between HLA-B27 misfolding and the production of proinflammatory cytokine production [43,44].
In a study about association between UPR activation and HLA-B27 related diseases in HLA-B27 transgenic rats, accumulation of misfolded B27 heavy chain was determined in splenocytes and macrophages from in those transgenic rats. Furthermore, UPR activation was demonstrated that it can be occurred in bone marrow macrophages from rats with active disease but not in younger animals that has not have the disease yet [45,46]. These studies were the first proof to explain association between increased expression of HLA-B27, misfolding and accumulation of its heavy chain in the ER, induction of UPR, and B27-associated disease phenotype.
In cells, levels of misfolded proteins are focused to monitor carefully and accumulation of misfolded proteins, for example in cytosol, induces a response to help protein refolding accumulated protein. Familiarly, in ER, increased level of transcription of genes that facilitates misfolded protein to refold involved in retrotranslocation and protein degradation in the cytosol [1]. The studies about protein quality process have been dealt with its increased complexity but the importance of relation between protein misfolding pathogenesis of a number of diseases [47]. Even in a full MHC class I assembly induction event, disulfide-linked homodimers and multimers can be formed as a result of accumulation of misfolded HLA-B27 heavy chains in the ER. Studies about B pocket mutants with developed folding specialties but still retaining Cys67 residue showed that any dimerization or accumulation have been occurred in ER; so this explains that covalent dimerization is a result of misfolding instead of a cause [25].
Among all factors that can be a key for understanding HLA-B27 misfolding and SpAs, CREBH, a transcription factor within the liver, is activated via elevated ER stress induction and the proinflammatory mediators and cytokines. Activated CREBH has enhanced production of proinflammatory mediators and also other transcription factors of the ER stress pathway can be participated in proinflammatory cytokine production such as XBP-1 and CHOP [48-50]. Proinflammatory cytokines, exclusively TNFα, can elevate ER stress and this event can cause a positive feedback mechanism that HLA-B27 misfolding lead to induce ER stress and proinflammatory cytokine production [51, 52]. That feedback mechanism can contribute to understand the link between ER stress and a proinflammatory cytokine environment, also characteristic feature of AS patients.
Interestingly, there may be a systematic relation between capacities to trigger UPR of HLA-B27 subtypes in terms of protein folding characteristics and AS disorder. According to recent data, B*2706 and B*2709 have much more folding capacity than disease-associated B*2702, B*2704 and B*2705 subtypes, indicating a synergy between folding properties, HLA-B27 subtypes and association to AS. Although B*2707 subtypes has the most prevalence among all subtypes in AS patients in most of the populations examined, it can be fold efficiently same with other non-disease associated subtypes. Therefore, relation between folding, HLA-B27 subtypes and disorder association is not directly proportional [53].
There is another key factor for understanding how HLA-B27 can cause disease which is production of transgenic rat models. Firstly, HLA-B27/human β2m (hβ2m) transgenic rats were demonstrated that those transgenic rats can manifest spontaneous spondyloarthritis-like phenotype that included colitis and arthritis [45]. This breakthrough firstly proved that HLA-B27 could directly cause disease and HLA-B27 can be associated with SpAs. Interestingly, CD8α/β T cells are not essential for arthritis or colitis manifestation. Also, spondyloarthritis-like phenotype from occurring in rats cannot be avoided with antibody-mediated CD8+ T-cell depletion and a chemically induced mutation in the CD8a gene. These findings are supported with that there will be no developing arthritis or colitis in CD4+ T cells efficiently transfer colitis to athymic nude rats if high level of HLA-B27 /hβ2m expression is carried on the bone marrow [38,54,55].
Despite of several studies about HLA-B27 transgenic rat strains, how some of these strains remain healthy is still mysterious; yet this enigma can be explained with the genetic backgrounds of the strains that were chosen for these studies [56]. Also, this effect can be related with 95–99% of HLA-B27 positive individuals remain healthy. It is possible that HLA-B27 overexpression may achieve the need for additional susceptibility alleles and enhance increased penetrant phenotypes. It has been considered that people with developing ankylosing spondylitis has an increased risk with homozygosity for HLA-B27 [57,58]. It has been never reported a copy number variation in HLA-B human locus. Therefore, studies must be focused on low copy number variations in HLA-B27 transgenic rats with genetic modifications for tendency in ankylosing spondylitis.
The frequency of HLA-B27 in patients with AS is approximately 94% [64]. In addition to HLA-B27, also HLA-B27 subtypes have been shown to have different incidences and relations with disease onset. According to previous data analyzing HLA-B27 polymorphisms and clinical symptoms related with spondyloarthropathies, no relations have been found between AS and HLA-B-27:06 or HLA [62,63]. Cytokines analysis is also a reliable marker inflammatory condition, comparing the cytokine profile of HLA-B27 positive and negative patients and healthy controls were performed by several studies. Since inflammation is an important component of AS, inflammation related cytokine profiles have gained importance. Rudwaleit et al. (2001) found that T cells producing TNF-α and IFN-γ are decreased in healthy controls both HLA-B27+ and HLA-B27- compared to HLA-B27+ AS patients [59]. Another interesting property of HLA-B27 is that β2-microglobulin free heavy chains of HLA-B27 were shown to interact with killer cell immunoglobulin-like receptor 3DL2 (KIR3DL2) and leucocyte immunoglobulin-like receptor B2 (LILRB2) with a higher affinity than heteromeric HLA-B27- β2-microglobulins in patients with spondyloarthropathies [60,61].
The researches to understand the molecular mechanisms of autoimmune diseases and ER stress-related diseases have been progressed tremendously in recent years. Several important achievements in this can be listed as the identification of the key players involved in these pathways and their organization to study how these pieces work together and organize the whole pathway.
Uncovering the crosstalk between ER-stress and HLA-B27 triggered autoimmune response and HLA-B27 misfolding; and also the intersections of ER-stress and/or HLA-B27 triggered autoimmune response with other cellular pathways will eventuate with important insights of cellular homeostasis.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, et al. (2008) Molecular Biology of the Cell. (5th edn). Garland Science, New York.
- Fawcet DW (1981) The Cell. (2nd edn). Saunders, Philedelphia.
- Loewy AG, Siekevitz P (1979) Cell Structure and Function. (3rd edn). Holt, New York.
- Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18: 716-731. [Crossref]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nature Rev Mol Cell Biol 8: 519-529. [Crossref]
- Schönthal AH (2012) Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica (Cairo) 2012: 857516. [Crossref]
- Gardner BM, Walter P (2011) Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333: 1891–1894. [Crossref]
- Shiu RPC, Pouyssegur J, Pastan I (1977) Glucose depletion accounts for the induction of two transformation sensitive membrane proteins in Rous sarcoma virus-transformed chick embryo fibroblasts. Proc Natl Acad Sci U S A 74: 3840-3844. [Crossref]
- Sanders SL, Whitfield KM, Vogel JP, Rose MD, Schekman RW (1992) Sec61p and BiP directly facilitate polypeptide translocation into the ER. Cell 69: 353-365. [Crossref]
- Caspersen C, Pedersen PS, Treiman M (2000) The sarco/endoplasmic reticulum calcium-ATPase 2ß is an endoplasmic reticulum stress-inducible protein. J Biol Chem 275: 22363-22372. [Crossref]
- Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13: 1211-1233. [Crossref]
- Lee AS (1992) Mammalian stress response: induction of the glucose-regulated protein family. Curr Opin Cell Biol 4: 267-273. [Crossref]
- Little E, Ramakrishnan M, Roy B, Gazit G, Lee AS (1994) The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Crit Rev Eukaryot Gene Expr 4: 1-18. [Crossref]
- Wooden SK, Lee AS (1992) Comparison of the genomic organizations of the rat grp78 and hsc73 gene and their evolutionary implications. DNA Seq 3: 41-48. [Crossref]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: 326-332. [Crossref]
- Berndt JD (2014) Preventing Stress with Sugar. Sci Signal 7: ec83.
- Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3: E255-263. [Crossref]
- Gerlach JQ, Sharma S, Leister KJ, Joshi L (2004) A Tight-Knit Group: Protein Glycosylation, Endoplasmic Reticulum Stress and the Unfolded Protein Response. Biochem Biophys Res Commun 325: 250-256.
- Yamaguchi S, Ishihara H, Tamura A, Yamada T, Takahashi R, et al. (2004) Endoplasmic reticulum stress and N-glycosylation modulate expression of WFS1 protein. Biochem Biophys Res Commun 325: 250-256. [Crossref]
- Jung T, Catalgol B, Grune T (2009) The proteasomal system. Mol Aspects Med 30: 191-296. [Crossref]
- Antoniou AN, Powis SJ, Elliott T (2003) Assembly and export of MHC class I peptide ligands. Curr Opin Immunol 15: 75-81. [Crossref]
- Antoniou AN, Lenart I, Guiliano DB (2011) Pathogenicity of Misfolded and Dimeric HLA-B27 Molecules. Int J Rheumatol 2011: 486856. [Crossref]
- Guiliano DB, Fussell H, Lenart I, Tsao E, Nesbeth D, et al. (2014) Endoplasmic Reticulum Degradation–Enhancing-Mannosidase–like Protein 1 Targets Misfolded HLA–B27 Dimers for Endoplasmic Reticulum–Associated Degradation. Arthritis Rheumatol 66: 2976–2988. [Crossref]
- Allen RL, O’Callaghan CA, McMichael AJ, Bowness P (1999) Cutting edge: HLA-b27 can form a novel ß-microglobulin-free heavy chain homodimer structure. J Immunol 162: 5045-5048. [Crossref]
- Dangoria NS, DeLay ML, Kingsbury DJ, Mear JP, Uchanska-Ziegler B, et al. (2002) HLAB27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem 277: 23459-23468. [Crossref]
- Jardetzky TS, Lane WS, Robinson RA, Madden DR, Wiley DC (1991) Identification of self peptides bound to purified HLA-B27. Nature 353: 326-329. [Crossref]
- Mear JP, Schreiber KL, Münz C, Zhu X, Stevanovic S, et al. (1999) Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol 163: 6665-6670. [Crossref]
- Magnacca A, Persiconi I, Nurzia E, Caristi S, Meloni F, et al. (2012) Characterization of a proteasome and TAP-independent presentation of intracellular epitopes by HLA-B27 molecules. J Biol Chem 287: 30358-30367. [Crossref]
- McNeil AJ, Yap PL, Gore SM, Brettle RP, McColl M, et al. (1996) Association of HLA types A1-B8-DR3 and B27 with rapid and slow progression of HIV disease. QJM 89: 177-185. [Crossref]
- Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg HC, et al. (2006) Dominant influence of an HLA-B27 restricted CD8Tcell response in mediating HCV clearance and evolution. Hepatology 43: 563–572. [Crossref]
2021 Copyright OAT. All rights reserv
- Schlosstein L, Terasaki PI, Bluestone R, Pearson CM (1973) High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med 288: 704-706. [Crossref]
- Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, et al. (1997) Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum 40: 1823-1828. [Crossref]
- Breban M, Miceli-Richard C, Zinovieva E, Monnet D, Said-Nahal R (2006) The genetics of spondyloerthropathies. Joint Bone Spine 73: 355-362. [Crossref]
- Caillat-Zucman S (2008) Molecular mechanisms of HLA association with autoimmune diseases. Tissue Antigens 73: 1-8. [Crossref]
- Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ (2004) Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem 279: 8895-8902. [Crossref]
- Tran TM, Satumtira N, Dorris ML, May E, Wang A, et al. (2004) HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol 172: 5110–5119. [Crossref]
- Bird LA, Peh CA, Kollnbeger S, Elliott T, McMichael AJ, et al. (2003) Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol 33: 748-759. [Crossref]
- Taurog JD, Maika SD, Satumtira N, Dorris ML, McLean IL, et al. (1999) Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev 169: 209-223. [Crossref]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332: 462-464. [Crossref]
- Welihinda AA, Tirasophon W, Kaufman RJ (1999) The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr 7: 293-300. [Crossref]
- Colbert RA (2000) HLA-B27 misfolding: a solution to the spondyloarthropathy conundrum? Mol Med Today 6: 224-230. [Crossref]
- Masters SL, Simon A, Aksentijevich I, Kastner DL (2009) Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 27: 621-668. [Crossref]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897-904. [Crossref]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, et al. (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2a. Mol Cell Biol 22: 7405-7416. [Crossref]
- Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD (1990) Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human 2m: an animal model of HLA-B27-associated human disorders. Cell 63: 1099-1112. [Crossref]
- Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, et al. (2005) HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol 175: 2438-2448. [Crossref]
- Colbert RA, Tran TM, Layh-Schmitt G (2014) HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol 57: 44-51. [Crossref]
- Goodall JC, Wu C, Zhang Y, McNeil L, Ellis L, et al. (2010) Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A 107: 17698-17703. [Crossref]
- Martinon F, Chen X, Lee AH, Glimcher LH (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11: 411-418. [Crossref]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, et al. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124: 587–599. [Crossref]
- Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH (2006) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1a-mediated NF-?B activation and down-regulation of TRAF2 expression. Mol Cell Biol 26: 3071–3084. [Crossref]
- Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, et al. (2005) Tumor necrosis factor a (TNFa) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFa. J Biol Chem 280: 33917–33925. [Crossref]
- López de Castro JA (2007) HLA-B27 and the pathogenesis of spondyloarthropathies. Immunol Lett 108: 27-33. [Crossref]
- May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, et al. (2003) CD8 alpha beta T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol 170: 1099-1105. [Crossref]
- Taurog JD, Dorris ML, Satumtira N, Tran TM, Sharma R, et al. (2009) Spondylarthritis in HLA-B27/human beta2-microglobulin-transgenic rats is not prevented by lack of CD8. Arthritis Rheum 60: 1977-1984. [Crossref]
- Weinreich S, Eulderink F, Capkova J, Pla M, Gaede K, et al. (1995) HLA-B27 as a relative risk factor in ankylosing enthesopathy in transgenic mice. Hum Immunol 42: 103-115. [Crossref]
- Jaakkola E, Herzberg I, Laiho K, Barnardo MC, Pointon JJ, et al. (2006) Finnish HLA studies confirm the increased risk conferred by HLA-B27 homozygosity in ankylosing spondylitis. Ann Rheum Dis 65: 775-780. [Crossref]
- Reveille JD (2012) Genetics of spondyloarthritis--beyond the MHC. Nat Rev Rheumatol 8: 296-304. [Crossref]
- Rudwaleit M, Siegert S, Yin Z, Eick J, Thiel A, et al. (2001) Low T cell production of TNFalpha and IFNgamma in ankylosing spondylitis: its relation to HLA-B27 and influence of the TNF-308 gene polymorphism. Ann Rheum Dis 60: 36-42. [Crossref]
- Raine T1, Brown D, Bowness P, Hill Gaston JS, Moffett A, et al. (2006) Consistent patterns of expression of HLA class I free heavy chains in healthy individuals and raised expression in spondyloarthropathy patients point to physiological and pathological roles. Rheumatology (Oxford) 45: 1338-1344. [Crossref]
- Giles J1, Shaw J, Piper C, Wong-Baeza I, McHugh K, et al. (2012) HLA-B27 Homodimers and Free H Chains Are Stronger Ligands for Leukocyte Ig-like Receptor B2 than Classical HLA Class 1. Immunol 188: 6184-6193. [Crossref]
- Taurog JD, Chhabra A, Colbert RA (2016) Ankylosing Spondylitis and Axial Spondyloarthritis. N Engl J Med 374: 2563-2574. [Crossref]
- Khan MA (2013) Polymorphism of HLA-B27: 105 subtypes currently known. Curr Rheumatol Rep 15: 362. [Crossref]
- Brown MA, Pile KD, Kennedy LG, Calin A, Darke C, et al. (1996) HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Ann Rheum Dis 55: 268-270. [Crossref]