Abstract
Cardiac arrhythmias are a major type of cardiovascular diseases and account for high morbidity and mortality. The occurrence of cardiac arrhythmias is closely associated with abnormal neurohumoral regulation of heart rhythmicity and the pathogenesis of many cardiovascular diseases, particularly coronary artery disease (CAD). Normal immunometaboliccoupling between endoplasmic reticulum (ER) and mitochondria is an essential condition for normal heart rhythm. ER stress -and mitochondrial oxidative stress evoked immunometabolic challenges can cause inflammation, lipotoxicity and cell apoptosis, leading to CAD and the ensuing arrhythmias. These disorders are associated with activation of inducible nitric oxide synthase, protein tyrosine nitration, malfunctioned chaperone, abnormal calcium signaling through mitochondria-associated ER membrane, disruption of oxidative phosphorylation, and activation of inflammatory and apoptotic pathways. In this review, we highlight the mechanisms underlying arrhythmias, specifically involving disorders in immuno-metabolic network and the underlying signaling process.
Key words
Arrhythmia, coronary artery disease, immunology, metabolism
Introduction
As a major type of cardiovascular disease (CVD), arrhythmias account for both high morbidity and mortality. In Europe and North America, as of 2014, atrial fibrillation (AF) affects about 2% to 3% of the population; approximately 80% of sudden cardiac death results from ventricular arrhythmias [1]. According to World Society of Arrhythmias, there are 20 million patients suffering from arrhythmias including 8 million with AF and 0.54 million with sudden cardiac death/year in 2015. Thus, understanding of the regulation of cardiac arrhythmias and developing more efficient therapies are critical.
Recently, the association of immunometabolic disorders with the occurrence of arrhythmias has emerged as a new horizon of studies; however, the underlying mechanism for immunometabolic disorder-associated arrhythmias remains poorly understood. In this paper, we review current evidence supporting the involvement of vicious cycle of immune-metabolic dysregulation in the pathogenesis of arrhythmias.
Cardiac arrhythmias and the etiology
Cardiac arrhythmia refers to irregular heart rhythm or rate due to abnormal generation or conduction of electrical impulses in the heart. The abnormal electrical activity in arrhythmias is commonly due to dysfunction and/or structural disruption of the electrical conduction system of the heart and can be classified into different types based on classifications of their pathogeneses.
Bradyarrhythmias
Bradyarrhythmias occur when action potentials fail to initiate or conduct appropriately in the rhythmic system including sinoatrial node, atrial-ventricular bundle, atrial ventricular node, and His-Purkinje fibers. Dysfunction in impulse initiation is usually due to slowed phase 4 of action potentials in the rhythmic cells [2,3] while the underlying mechanisms are largely determined by the sources of pathological changes.
Extrinsic causes of bradyarrhythmias mainly include medications, hypothyroidism, sleep apnea, and increased intracranial pressure. In rare cases, vagal hyperactivity can also result in bradyarrhythmias, even cardiac arrest. Some of these causes maybe reversible such as hypoxia, hypothermia, and those that are induced by medications [4]. In contrast, the intrinsic causes are mainly related to degenerative processes such as post-infarction myocardial fibrosis of the sinoatrial node or the conduction system, and inflammatory diseases including pericarditis, rheumatic cardiomyopathy and myocarditis [5,6].
Tachyarrhythmias
Tachyarrhythmias can result from 1) enhanced automaticity; 2) re-entry arrhythmia or, 3) triggered arrhythmia. In arrhythmias caused by enhanced automaticity, it is essential for the acceleration of spontaneous depolarization of pacemaker cells in association with shortening of ventricular repolarization and refractoriness [5], which often results from the activation of sympathetic activity from both the central and peripheral approaches [6].
When arrhythmias are associated with cardiac ischemia/reperfusion (I/R) injury, abnormal automaticity can lead to atrial and ventricular tachyarrhythmias, likely due to propensity of ischemic tissue to depolarize neighboring non-ischemic, healthy myocardium through either anterograde or retrograde limbs, in which re-entry arrhythmia is the most common form of tachyarrhythmia. Such arrhythmia is clinically associated with atrial ventricular re-entry, atrial flutter, and ventricular arrhythmia in post-ischemic myocardium [7].
Triggered arrhythmia occurs during after-depolarization of the action potential and frequently under cardiac ischemia and digitalis toxicity. After ischemic injury, surviving cells could spontaneously release Ca2+ from sarcoplasmic reticulum to evoke arrhythmias [8]through multiple approaches, such as changes in the production of microRNAs [9].
Many CVDs predispose the heart to arrhythmias, such as inflammatory cardiomyopathy, I/R injury during myocardial infarction, and metabolic CVDs [7]. Thus, the occurrence of arrhythmias is not only involving neurohumoral regulation of the rhythmic system, but also cardiac pathology. In this review, we focus on the immunometabolic basis of arrhythmias in the following section.
Immunometabolic basis of arrhythmias
In the pathogenesis of these CVDs, immunologic injury and metabolic disorders are essential factors that constitute the immunometabolic basis of cardiac arrhythmias.
Immunological injury and arrhythmias
The heart shares with other organs the susceptibility to immunological injury. Many CVDs are characterized by the presence of inflammatory cells within the myocardium and autoantibodies in the sera due to immune sensitization to endogenous or exogenous cardiac antigens.
Humoral immunologic injury
The immunopathogenesis of cardiac rhythm and conduction disorders have been identified in sick sinus syndrome, bradyarrhythmias, and hypersensitive carotid sinus syndrome. For instance, cardiac autoantibodies could be detected in the sera of patients following infarction and/or cardiotomy [10].Now it has been commonly accepted that the humoral immunological injuries are related to autoimmune disorders and non-specific humoral factors[11-13]. These facts support the proposal that autoimmunity and inflammation take part in electrical and structural remodeling of left atrium and predispose patients with autoimmune and inflammatory diseases to increased risk of arrhythmia [14].
Cellular immunologic etiology
The cellular type of immunological injury is associated with the activation of cytotoxic T-lymphocytes/cells. This has been well demonstrated in studies on the association of T cell-mediated cytotoxicity and the electric instability in those with long QT syndrome and catecholaminergic polymorphic ventricular tachycardia [15]. In a mouse model of chronic ischemic heart failure, there were global expansion and activation of CD4+ T cells to induce cardiac injury and remodeling [16]. It was also reported that there are plenty of mast cells in the heart, and they are predominantly located around the coronary adventitia and in close contact with small vessels in the muscle wall. The release of mediators can influence ventricular function, heart rate, and coronary artery tone, potentially triggering arrhythmias [17]. These evidences indicate that aberrant immune reactions can cause heart injuries through both humoral and cellular approaches; correspondingly restoration of immunologic homeostasis could potentially protect the heart from arrhythmias.
Metabolic disorders and arrhythmias
Normal metabolism of the cardiovascular system is the prerequisite of normal HR. Varieties of metabolic disorders such as high triglyceride, low high-density lipoprotein and obesity as well as high or low plasma glucose levels and diabetes are associated with increased risk of atherosclerosis, CAD and the resultant arrhythmias.
Dyslipidemia and lipotoxicity
Dyslipidemia in association with abnormal amounts of cholesterol, triglycerides and unsaturated fatty acids in blood increases risk of arrhythmias. This has been verified by a clinical investigation of 35232 patients in Sweden revealed that AF in humans had substantially greater proportion of polyunsaturated diacylglycerols in the cardiac tissues [18]. Low high-density lipoprotein, a hallmark of metabolic syndrome, is closely related to AF risks by slowing atrial conduction as shown by the prolongation of P wave, PR intervals, QRS duration and corrected QT interval in mice [19]. Furthermore, very-low-density lipoproteins induced downregulation of connexin (Cx)-40 and Cx-43 at transcriptional, translational and tissue levels but impaired stability of gap junctions [19]. These facts indicate that dyslipidemia predisposes arrhythmias through cardiac lipotoxicity.
Glucose metabolism disorders
Similar to dyslipidemia, glucose metabolism disorders also promote the occurrence of arrhythmias, which is represented by diabetes cardiomyopathy and I/R heart. For example, glucose ingestion increased QT interval and aggravated the cardiac repolarization disturbances in long-QT syndrome type 2 patients [20]. Hyperglycemia can inhibit myocardial anesthetic post-conditioning, and result in oxidative stress and apoptosis in I/R condition [21]. In addition, glycated hemoglobin is a long-term measure of glucose control and has also association with AF risk [22]. Thus, both defective glucose metabolism and low tissue oxygenation may contribute to arrhythmias after acute myocardial infarction in diabetic patients. It is worth noting that reduced levels of glucose or hypoglycemia in type 2 diabetes mellitus (T2DM) can also cause arrhythmias [23]. Moreover, individuals with T2DM demonstrate greater repolarization abnormalities for a given hypoglycemic stimulus, which is related to sympathoadrenal stimulation and hypokalemia [24].
Signaling pathway mediating immunometabolic disorder-associated arrhythmias
The effect of immunometabolic disorders on atherosclerosis and the subsequent arrhythmias is closely associated with inactivation of the reperfusion injury salvage kinase (RISK) pathway, AMP-activated protein kinase (AMPK) and protein kinase C (PKC) signaling.
RISK pathway
The RISK pathway including phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt)-endothelial nitric oxide (NO) synthase (eNOS) cascades and extracellular signal-regulated protein kinase (ERK) 1/2 has been extensively identified in the cardioprotective effects of normal immunometabolic activity. This fact has been confirmed by studies on the cardioprotective effect of ginseng extract in rat heart [25] and estradiol-17βtreatment in high fat-fed rats [26].
In the RISK pathway, NO production is a key step in cardioprotection against myocardiac I/R injury. However, NO from the eNOS but not inducible NOS (iNOS) exerts the protective effect. For example, the protective effect of adipocyte fatty-acid-binding protein deficiency did not occur in eNOS (-/-) mice but occurred in iNOS (-/-) and wild type mice [27], supporting that eNOS is required for its cardioprotection. By contrast, the PI3K/Akt signaling pathway activated by estradiol- 17β can attenuate the detrimental effects of increased iNOS expression/activity by inhibiting nitrosylation of functional proteins [26].
A key downstream target of eNOS/NO in cardioprotection is the mitochondrial ATP-dependent potassium (mKATP) channels. This has been supported by the findings of myocardial protection by hydrogen sulfate (H2S) [28] and isoflurane-induced myocardial postconditioning under acute hyperglycemia [21]. Among them, the effect of H2S is highly representative.H2S is mainly converted from cyst(e)ine catalyzed by cystathionineγ-lyase that is activated by Ca2+-calmodulin and PI3K. Downstream targets of the H2S include PI3K and ERK ½ [29]. Thus, H2S can not only respond to mobilization of Ca2+-calmodulin signaling, but also activate RISK pathway to protect the heart through multiple layers, such as antioxidative action, preservation of mitochondrial function, reduction of apoptosis, anti-inflammatory responses, angiogenic actions, regulation of mKATP channel, and interaction with NO.
AMPK pathway
In parallel with the RISK pathway, AMPK signaling could also been an upstream enzyme of the Akt-NO pathway. This proposal is supported by the studies on vascular endothelial cells in mice [30] and human umbilical vein endothelial cells [31]. By contrast, the AMPK activity is decreased in obesity and T2DM and increased with metabolically favorable conditions and cholesterol lowering drugs [32].
AMPK can inhibit pro-inflammatory reactions through multiple approaches. For example, AMPK activation inhibited interleukin (IL)-1β-stimulated CXCL10 secretion, reduced IL-1 receptor-associated kinase-4 phosphorylation and downregulated MKK4/ c-Jun N-terminal kinase (JNK) and IKK/IkappaB/ nuclear factor-kappaB (NF-kB) signaling [33]. In addition, AMPK can inhibit HMG-CoA reductase to reduce cholesterol synthesis and reduce inflammation [34]. Thus, the inhibition of multiple pro-inflammatory signaling pathways by AMPK can underlie the immunometabolic protection of the HR.
PKC signaling
A critical signaling event in cardioprotection is the activation of PKC. Chelerythrine, a PKC enzyme inhibitor, can block the cardioprotective effects of oxytocin in reducing infarct size, plasma levels of creatine kinase-MB and lactate dehydrogenase as well as the severity and incidence of ventricular arrhythmias in I/R rats [35]. However, different isoforms of PKC may have different, even opposite effects.
PKC-εin the mitochondria is known as a modulator of cardiac cell metabolism through activation of the mKATP channelfollowing cGMP production and subsequent activation of protein kinase G. In cardioprotection, PKC-ε, likely a downstream effector of PLC-βand NO generated by eNOS, has been shown in antioxidant-increased Cx-43 and suppression of ventricular fibrillation in rats [36]. In contrast, activation of PKC-α, a downstream event of PLC-β1b that is a heart-specific signaling, leads to cardiac injury. As reported, hypoxic activation of PKC-αdramatically increased iNOS expression, concomitant to enhanced apoptotic cell percentage and molecular interaction between apoptotic protease activating factor-1 and cytochrome (Cyt) C [37]. These findings indicate that PLC-β1b-PKC-α-iNOS signaling pathway mediates the activation of mitochondrial apoptotic pathway.
Interaction between immunological and metabolic processes
In the pathogenesis of CVDs and the resultant arrhythmias, immunologic injuries and metabolic disorders occur simultaneously and are intertwined to form a malfunctioned immunometabolic network.
Metabolic influence of immune activity
Atherosclerosis is a chronic inflammatory disease that is mediated by innate and adaptive immune responses; the immune responses could be evoked by abnormal metabolic activity. As reported, high levels of circulating fatty acids and triglycerides can evoke inflammatory signaling in the heart with increased reactive oxygen species (ROS) and NF-kB activity, mitochondrial dysfunction and insulin resistance [38]. Exposure to saturated fatty acids, such as palmitate, led to ER stress in macrophages and promotes inflammation in atherosclerotic plaques [39]. Diabetic cardiomyopathy showed oxidative stress and apoptosis in cardiac cells in rats, which could be reduced by multiple antioxidants that reduced levels of lipid peroxidation and levels of pro-inflammatory transcription factor NF-kB as well as cytokines such as tumor necrosis factor-α(TNF-α), interferon-γ, TGF-β, and IL-10 [40]. Moreover, activated simvastatin (HMG-CoA reductase inhibitor) infusion prior to reperfusion in a pig model of I/R reduced coronary and cardiac oxidative DNA-damage, diminished neutrophil infiltration at the site of ischemia but preserved mitochondrial membrane potential [DeltaPsi(m)] and reduced apoptosis in the ischemic myocardium [34]. It was also reported that sub-endothelial accumulation and modification of lipids in the artery wall trigger an inflammatory reaction in mice. This reaction can be weakened by regulatory T cells that suppress T cell proliferation and secretion of anti-inflammatory cytokines (e.g. IL-10 and transforming growth factor-β) [41]. In addition, some polysaccharide fractions was found to stimulate the production of NO and immunomodulatory cytokines (IL-1βand TNF-α) without cytotoxicity in macrophages [42].
Immunologic influence on metabolism
On the other hand, immune reactions can cause metabolic disorders. This has been well demonstrated in studies on obesity that is generally considered a CVD-associated metabolic/inflammatory disease. In adipose tissue of high fat-fed animals, innate and adaptive immune cell responses play critical roles in the regulation of metabolic homeostasis. In the lean state, type 2 cytokine-associated immune cell responses predominate in white adipose tissue and protect against weight gain and insulin resistance through direct effects on adipocytes and elicitation of beige adipose. In obesity, these metabolically beneficial immune pathways become dysregulated, and adipocytes and other factors initiate metabolically deleterious type 1 inflammation that impairs glucose metabolism [43]. It was also reported that low-grade systemic inflammation associated with obesity leads to cardiovascular complications, caused partly by infiltration of adipose and vascular tissue by effector T cells. This change was mediated via direct exposure of CD4+ T cells to palmitate [44]. Clearly, metabolic disorders in the heart can result from immunologic injuries.
Together with a previous review [45], the facts presented above allow us to propose that accumulation of harmful lipids or generation of signaling intermediates can interfere with immune regulation in cardiac tissues and vice versus; they can form a vicious cycle of immune-metabolic dysregulation and in turn hurt cardiac functions. Thus, in studying on the pathogenesis of arrhythmias and therapeutic approaches, the immune states and metabolic conditions should be considered in an intact immunometabolic network.
Roles of ER and mitochondrion in arrhythmia-associated cellular injuries
Immunometabolic disorders involve dysregulation of multiple organelles during cardiac injuries and the ensuing arrhythmia. ER stress and mitochondrial oxidative stress intersect immune injury and metabolic disorder, thereby highly representing malfunctions of immunometabolic network in arrhythmogenesis.
ER stress
The ER is responsible for the folding, processing and trafficking of all secretory and integral membrane proteins to the cell surface. It is also a pivotal site for the quality control of proteins, Ca2+ homeostasis, and biosynthesis of cholesterol and lipids. Thus, abnormal Ca2+ regulation, viral infection, high-fat diet, hypoglycemia, and lack of amino acids can all disrupt ER homeostasis and cause the accumulation of unfolded proteins, leading to ER stress and disruption of ER integrity. For example, palmitate-induced downregulation of sorcin, a calcium binding protein, increased levels of glucose-6-phosphatase catalytic subunit-2, which is a negative regulator of glucose-stimulated insulin secretion, leading to ER stress [46]. In contrast, statins can reduce ER stress by inhibiting cholesterol synthesis in the ER and alleviate myocardial infarction [34].
Examples of the immunometabolic disorders have been well documented in studies on saturated fatty acids-induced ER stress. Endogenous H2S levels in serum of diabetic cardiomyopathy patients and rats, and its contents in rat heart were significantly lower, which was accompanied with cardiac lipotoxicity and the process was inhibited by ER stress inhibitor or H2S [47].In another example, homocysteine-evoked myocardial fibrosis can be inhibited by a paracrine/autocrine peptide intermedin1-53 in apolipoprotein E-deficient mice and neonatal rat cardiac fibroblasts.This effect was accompanied with inhibition of the upregulation of inositol-requiring enzyme-1, an ER stress marker and inflammatory factor (e.g. TNF-α, monocyte chemotactic protein-1, IL-6, and IL-1β). In addition, the ER stress is also associated with rapamycin complex 1 (mTORC1) signaling [48] and its associated chaperone processes [49].
It is important to note that ER stress can propagate through inactivation of inositol-requiring enzyme-1, the key ER regulator, through nitrosylation due to excessively produced NO following increased iNOS activity [50]. This is induced by dyslipidemia-associated activation of poly(ADP-ribose) polymerase-1 in vascular dysfunction, which leads to expansion of initial event in the heart to affect the rhythmic system, resulting in arrhythmias. Together with the evidences reviewed recently[45] that ER is a critical organelle mediating both metabolic and inflammatory adaptive responses to proteotoxic, nutritional, and energy-related stresses, we propose that ER is an important immunometabolic hub in the cardiac pathogenesis and the ensuing arrhythmias.
Mitochondrial oxidative stress
Significant increase in ROS production is a primary cause of cardiomyocyte apoptosis in diabetic cardiomyopathy as well as I/R injury to the heart. ROS is a natural byproduct of the normal metabolism of oxygen including peroxides, superoxide, hydroxyl radical, and singlet oxygen. In diabetic cardiomyopathy and I/R, cardiac production of ROS can increase dramatically, resulting in oxidative stress and significant cellular damage. The process occurs primarily in mitochondriawhen oxygen is prematurely and incompletely reduced during oxidative stress and superoxide anion is generated, leading to destruction of the mitochondria and cell apoptosis through Bcl-2/Bax-Cyt C/apoptotic protease activating factor 1-caspase-9 pathway [51].
Mitochondrial ROS formation during reperfusion is based on the opening of mitochondrial permeability transition pores (MPTP) and the resultant calcein release, DeltaPsi(m) collapse, and disturbance of ATP recovery in isolated cardiac myocytes from adult I/R rats. During the reperfusion, Ca2+ oscillations occurred, mitochondrial Ca2+ concentration and mitochondrial ROS levels increased, cells developed hypercontracture and underwent necrosis. Suppression of the mitochondrial Ca2+ uptake or MPTP opening significantly attenuated Ca2+ oscillation, hypercontracture and necrosis. These changes were not influenced by reduced mitochondrial ROS levels since ROS scavengers had no effect on these parameters [52]. Thus, the interplay between Ca2+ oscillation and MPTP promotes the reperfusion-induced cardiomyocyte injury. It remains to differentiate the source of mitochondrial Ca2+ contributed from sarcoplasmic reticulum and from the ER. However, I/R-evoked Ca2+ oscillation can cause cardiac injury through mitochondrial Ca2+ overload and the resultant cardiomyocyte apoptosis [53].
It is worth noting that increased ROS level has dual effect on the heart, largely determined by its extent and duration. In response to sudden I/R insults, there was a burst increase in ROS production, which caused dramatic oxidative stress and myocardiac injury. However, the production of ROS evoked by protective agents could protect the heart by prevention of I/R-evoked ROS burst, thereby exerting the antiarrhythmic function. For example, isoflurane upregulated ROS generation before but inhibited a ROS burst after challenge []. Consistent finding is also present in H2O2-induced cardioprotection against reperfusion injury. As reported in the Langendorff-perfused rat hearts, pretreatment of H2O2 significantly improved the post-ischemic recoveries in intracellular phosphocreatine and ATP levels. In isolated permeabilized myocytes, H2O2 accelerated the calcein leakage from mitochondria by opening the MPTP. However, H2O2 did not depolarize DeltaPsi(m) even in the presence of ATPase but decreased mitochondrial Ca2+ concentration by accelerating the mitochondrial Ca2+ extrusion via an MPTP []. It is likely that stabilization of the DeltaPsi(m) is critically important for maintaining normal oxidative phosphorylation and immunometabolic balance even though mild ROS challenge partially increased the leakage through MPTP and decreased mitochondrial Ca2+ levels.
ER-mitochondrial couplings in immunometabolic regulation
It is also important to note that there are close interactions between ER and mitochondrion in of cardiac activity through the mitochondria-associated ER membrane (MAM) and associated proteins. MAMs in the heart are responsible for Ca2+ signaling between ER and mitochondria [56]. This Ca2+ signaling is regulated by glycogen synthase kinase-3β(GSK3β) protein in the ER and MAMs in the heart. Phosphorylation/activation of GSK-3βoccurs following activation of JNK induced by advanced glycation end-products in diverse pathological settings including diabetes, inflammation and acute I/R injury in the heart. GSK3βspecifically interacted with the inositol 1,4,5-trisphosphate receptors (IP3Rs) Ca2+ channeling complex in MAMs. During I/R, increased GSK3βleads to enhanced transfer of Ca2+ from ER to mitochondria, leading cytosolic and mitochondrial Ca2+ overload and subsequent cell death. Inhibition of GSK3βat reperfusion reduced both IP3R phosphorylation and ER Ca2+ release, which consequently diminished both cytosolic and mitochondrial Ca2+ concentrations as well as sensitivity to apoptosis [57]. This finding is in agreement with a previous report that inactivation of GSK-3βdirectly or indirectly by mKATP channel activation facilitates recovery of DeltaPsim by suppressing ROS production and mPTP opening, leading to cytoprotection from oxidant stress-induced cell death. That is, acceleration of the DeltaPsim recovery contributes to cytoprotection afforded by activation of the mKATP channel or inactivation of GSK-3β. In addition, in H9c2 cells, opening of the MPTP with antimycin reduced DeltaPsim and induced calcein leak from mitochondria, which were partially recovered by mKATP channel openers. Activation of the mKATP channel induced inhibitory phosphorylation of GSK-3βand suppressed ROS production, LDH release and apoptosis after antimycinwashout [58-60].
Another key molecule bridging the mitochondria and ER is mitofusin-2 (Mfn-2), a mitochondrial fusion protein known to be critical in regulating cardiac function. As reported, angiotensin II and miR-106a treatment cultured cardiomyocytes mitochondria presented cristae defects with considerable depolarization of mitochondrial membrane and increased ROS production, which was reversed by overexpression of Mfn2, a function opposite to GSK3β [9]. Additionally, hyperglycemia stimulated ER interactions and mitochondrial apoptotic pathways in cardiac tissues of diabetic rats. The processes were inhibited by exogenous H2S treatment through the regulation of Mfn-2 expression in association with inhibition of the interaction between the ER and mitochondrial apoptotic pathway, leading to decrease in the expression of mitochondrial apoptotic proteins, Cyt C, and MPTP opening [42]. These findings indicate that ER and mitochondria function like an “organelle syncytium” in the immunometabolic regulation of cardiac activity through histological and functional couplings and under the close regulation of antiarrhythmic signaling events. Figure 1 is a diagrammatic illustration of the hypothetical mechanism of immunometabolic disorders-elicited arrhythmias.
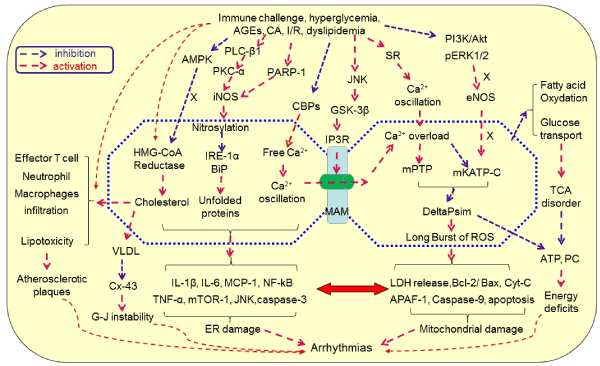
Figure 1.Hypothetical mechanism of immunometabolic disorders-elicited arrhythmias.Immunological challenges and metabolic disorders can activate inflammation, induce endoplasmic reticulum (ER) stress, inhibit UPR and chaperone, increase the production of cholesterol and VLDL, promote the production of immune cytokine, increase ER-mitochondrial Ca2+ signaling through MAMs and Ca2+ overload, activate mitochondrial oxidative stress, disrupt TCA cycle, and activate apoptotic pathway, leading to degeneration and cell death. These processes are signaling through activating iNOS, PKC-α, JNK and GSK-3β while inhibiting AMPK, CBPs, PI3L/Akt, pERK1/2 and eNOS. As a result, lipotoxicity, atherosclerosis and CAD occur, leading to arrhythmias once the rhythmic system is involved directly or indirectly. Abbreviations: AGEs, advanced glycation end-products; AF, atrial fibrillation; Akt, protein kinase B; AMPK, AMP-activated protein kinase; APAF-1, apoptotic protease activating facter-1; Bcl-2, B-cell lymphoma-2 protein; BiP, binding immunoglobulin protein; CA, catecholamine; CAD, coronary artery disease; CBPs, calcium binding proteins; CGL, cystathionine gamma-lyase; CVD, cardiovascular disease; Cx-43, connexin-43; Cyt C, cytochrome c; DeltaPsi(m), mitochondrial membrane potential; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ERK1/2, extracellular signal-regulated protein kinase 1/2; G-J, gap junction; H2S, hydrogen sulfate; GSK3β, glycogen synthase kinase-3β; IL, interleukin; iNOS, inducible nitric oxide synthase; IP3Rs, inositol 1,4,5-trisphosphate receptors; I/R, ischemia/reperfusion; IRE-1, inositol requiring enzyme1; JNK, c-Jun N-terminal kinase; LDH, Lactate dehydrogenase; MAM, mitochondria-associated ER membrane; mATP C, mitochondrial ATP-dependent potassium channels; MCP-1, monocyte chemotactic protein-1; MPTP, mitochondrial permeability transition pores; PARP-1, poly(ADP-ribose) polymerase-1; PC, phosphocreatine; PI3K, phosphatidylinositol 3-kinases; PKC, protein kinase C; PLC, Phospholipase C; RISK, reperfusion injury salvage kinase; ROS, reactive oxygen species; TCA cycle, tricarboxylic acid cycle; TNF, tumor necrosis factor; UPR, unfolded protein response; VLDL, low-density lipoproteins.
Conclusion
Immune injuries and metabolic disorders are the central risk factors for CAD, but our current knowledge of the mechanisms underlying arrhythmias is still insufficient. Full understandings of the mechanisms underlying immunometabolic disorder-associated arrhythmias, particularly vicious cycle of immune-metabolic dysregulation, will help for designing more targeted therapies to block the pathogenesis of arrhythmias by suppressing ER stress and mitochondrial oxidative stress, thereby greatly reducing the incidence of arrhythmias and the mortality from CVDs.
References
- Zoni-Berisso M, Lercari F, Carazza T, Domenicucci S (2014) Epidemiology of atrial fibrillation: European perspective. Clin Epidemiol 6: 213-220.[Crossref]
- Motloch LJ, Ishikawa K, Xie C, Hu J, Aguero J, et al. (2017) Increased afterload following myocardial infarction promotes conduction-dependent arrhythmias that are unmasked by hypokalemia. JACC Basic Transl Sci 2: 258-269.[Crossref]
- Walraven G (2016) Basic Arrhymias, 8th ed. Pearson.
- Margulescu AD, Mont L (2017) Persistent atrial fibrillation vs paroxysmal atrial fibrillation: differences in management. Expert Rev Cardiovasc Ther 5:601-618. [Crossref]
- Osadchii OE (2017) Role of abnormal repolarization in the mechanism of cardiac arrhythmia. Acta Physiol (Oxf) 220 Suppl 712: 1-71.[Crossref]
- Yamaguchi K (2013) Tachycardic responses to stimulation of β-adrenoceptors in the brain parenchyma in conscious rats. Neurosci Res 76: 213-223.[Crossref]
- Romero J, Kumar S, Akira F, Briceño DF, Tedrow UB, et al. (2017) Emergence of Atrioventricular Nodal Reentry Tachycardia After Surgical or Catheter Ablation for Atrial Fibrillation: Are We Creating the Arrhythmia Substrate? Heart Rhythm piiS1547-5271(17)30963-3. [Crossref]
- Dobrev D, Wehrens XHT (2017) Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol595: 4001-4008.[Crossref]
- Guan X, Wang L, Liu Z, Guo X, Jiang Y, et al. (2016) miR-106a promotes cardiac hypertrophy by targeting mitofusin 2. J Mol Cell Cardiol99: 207-217.[Crossref]
- Friedman H, Specter S, Cerdan A, Cerdan C, Chang K (1983) Cell-mediated immune injury to the heart. Advances in experimental medicine and biology 161:479-489. [Crossref]
- Satta N, Vuilleumier N (2015) Auto-antibodies as possible markers and mediators of ischemic, dilated, and rhythmic cardiopathies. Curr Drug Targets 16: 342-360.[Crossref]
- Liu X, Zhang Y, Du W, Liang H, He H, et al. (2016) MiR-223-3p as a Novel MicroRNA Regulator of Expression of Voltage-Gated K+ Channel Kv4.2 in Acute Myocardial Infarction. Cell PhysiolBiochem 39:102-114. [Crossref]
- Ainsworth HC, Marion MC, Bertero T, Brucato A, Cimaz R, et al. (2017) Association of Natural Killer Cell Ligand Polymorphism HLA-C Asn80Lys With the Development of Anti-SSA/Ro-Associated Congenital Heart Block. Arthritis Rheumatol69: 2170-2174. [Crossref]
- Galloway A, Li H, Vanderlinde-Wood M, Khan M, Benbrook A, et al. (2015) Activating autoantibodies to the beta1/2-adrenergic and M2 muscarinic receptors associate with atrial tachyarrhythmias in patients with hyperthyroidism. Endocrine 49:457-463. [Crossref]
- Rizzo S, Basso C, Troost D, et al. (2014) T-cell-mediated inflammatory activity in the stellate ganglia of patients with ion-channel disease and severe ventricular arrhythmias. CircArrhythmElectrophysiol 7:224-229. [Crossref]
- Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, et al. (2017) Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail 10: e003688.[Crossref]
- Rojas-Perez-Ezquerra P, Noguerado-Mellado B, Morales-Cabeza C, Zambrano Ibarra G2, DatinoRomaniega T3 (2017) Atrial Fibrillation in Anaphylaxis. Am J Med 130: 1114-1116.[Crossref]
- Gizurarson S, Ståhlman M, Jeppsson A, Shao Y, Redfors B, et al. (2015). Atrial fibrillation in patients admitted to coronary care units in western Sweden - focus on obesity and lipotoxicity. J Electrocardiol48:853-860. [Crossref]
- Lee HC et al. (2017) Very-Low-Density Lipoprotein of Metabolic Syndrome Modulates Gap Junctions and Slows Cardiac Conduction. Sci Rep 7: 12050.[Crossref]
- Hyltén-Cavallius L, Iepsen EW, WewerAlbrechtsen NJ, Svendstrup M, Lubberding AF, et al. (2017) Patients With Long-QT Syndrome Caused by Impaired hERG-Encoded Kv11.1 Potassium Channel Have Exaggerated Endocrine Pancreatic and Incretin Function Associated With Reactive Hypoglycemia. Circulation 135: 1705-1719.[Crossref]
- Raphael J, Gozal Y, Navot N, Zuo Z (2015) Activation of Adenosine Triphosphate-regulated Potassium Channels during Reperfusion Restores Isoflurane Postconditioning-induced Cardiac Protection in Acutely Hyperglycemic Rabbits. Anesthesiology 122:1299-1311. [Crossref]
- Qi W, Zhang N, Korantzopoulos P, Letsas KP, et al. (2017) Serum glycated hemoglobin level as a predictor of atrial fibrillation: A systematic review with meta-analysis and meta-regression. PLoS One 12: e0170955.[Crossref]
- Reno CM, VanderWeele J, Bayles J, Litvin M, Skinner A, et al. (2017) Severe Hypoglycemia-Induced Fatal Cardiac Arrhythmias Are Augmented by Diabetes and Attenuated by Recurrent Hypoglycemia. Diabetes 66: 3091-3097.[Crossref]
- Novodvorsky P, Bernjak A, Chow E, et al. (2017) Diurnal Differences in Risk of Cardiac Arrhythmias During Spontaneous Hypoglycemia in Young People With Type 1 Diabetes. Diabetes Care 40: 655-662.[Crossref]
- Zhou M, Bao Y, Li H (2015) Deficiency of adipocyte fatty-acid-binding protein alleviates myocardial ischaemia/reperfusion injury and diabetes-induced cardiac dysfunction. Clin Sci (Lond) 129:547-559. [Crossref]
- Zafirovic S, Obradovic M, Sudar-Milovanovic E, Jovanovic A4, Stanimirovic J5, et al. (2017) 17β-Estradiol protects against the effects of a high fat diet on cardiac glucose, lipid and nitric oxide metabolism in rats. Mol Cell Endocrinol 446: 12-20.[Crossref]
- Zhou H, Hou SZ, Luo P (2011) Ginseng protects rodent hearts from acute myocardial ischemia-reperfusion injury through GR/ER-activated RISK pathway in an endothelial NOS-dependent mechanism. J Ethnopharmacol 135:287-298. [Crossref]
- Jin S, Teng X, Xiao L, Xue H, Guo Q, et al. (2017) Hydrogen sulfide ameliorated L-NAME-induced hypertensive heart disease by the Akt/eNOS/NO pathway. ExpBiol Med (Maywood) 242: 1831-1841.[Crossref]
- You X, Chen Z, Zhao H, Xu C, et al. (2017) Endogenous hydrogen sulfide contributes to uterine quiescence during pregnancy. Reproduction 153: 535-543.[Crossref]
- Chen Z, Peng IC, Sun W, Su MI, Hsu PH, et al. (2009) AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res 104: 496-505.[Crossref]
- Huang LY, Yen IC, Tsai WC, Ahmetaj-Shala B, et al. (2017) Rhodiolacrenulata Attenuates High Glucose Induced Endothelial Dysfunction in Human Umbilical Vein Endothelial Cells. Am J Chin Med 45: 1201-1216.[Crossref]
- Gujar AD, Ibrahim BA, Tamrakar P, Cherian AK, Briski KP (2014) Hindbrain lactostasis regulates hypothalamic AMPK activity and metabolic neurotransmitter mRNA and protein responses to hypoglycemia. Am J PhysiolRegulIntegr Comp Physiol 306:R457-469. [Crossref]
- Mancini SJ, White AD, Bijland S, Rutherford C, Graham D, et al. (2017) Activation of AMP-activated protein kinase rapidly suppresses multiple pro-inflammatory pathways in adipocytes including IL-1 receptor-associated kinase-4 phosphorylation. Mol Cell Endocrinol 440: 44-56.[Crossref]
- Vilahur G, Casani L, Pena E, et al.: 2014. HMG-CoA reductase inhibition prior reperfusion improves reparative fibrosis post-myocardial infarction in a preclinical experimental model. Int J Cardiol 175:528-538. [Crossref]
- Faghihi M, Alizadeh AM, Khori V, Latifpour M, Khodayari S (2012) The role of nitric oxide, reactive oxygen species, and protein kinase C in oxytocin-induced cardioprotection in ischemic rat heart. Peptides 37:314-319. [Crossref]
- BaÄovà BS, Vinczenzovà C, Åurmanovà J, KaÅ¡parovÃD, et al. (2017) Altered thyroid status affects myocardial expression of connexin-43 and susceptibility of rat heart to malignant arrhythmias that can be partially normalized by red palm oil intake. Histochem Cell Biol147: 63-73.[Crossref]
- Qiu Z, Zhang W, Fan F (2012) Rosuvastatin-attenuated heart failure in aged spontaneously hypertensive rats via PKCalpha/beta2 signal pathway. J Cell Mol Med 16:3052-3061.
- Dirkx E, Schwenk RW, Glatz JF, Luiken JJ, van Eys GJ: 2011. High fat diet induced diabetic cardiomyopathy. Prostaglandins, leukotrienes, and essential fatty acids 85:219-225. [Crossref]
- Pircher A, Treps L, Bodrug N, Carmeliet P (2016) Endothelial cell metabolism: A novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis 253: 247-257.[Crossref]
- Kumar S, Prasad S, Sitasawad SL (2013) Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocin-induced diabetic rats. PLoS One 8: e67009.[Crossref]
- Spitz C, Winkels H, Bürger C, Weber C, Lutgens E, et al. (2016) Regulatory T cells in atherosclerosis: critical immune regulatory function and therapeutic potential. Cell Mol Life Sci 73:901-922. [Crossref]
- Yang F, Yu X, Li T, Wu J, Zhao Y, et al. (2017) Exogenous H2S regulates endoplasmic reticulum-mitochondria cross-talk to inhibit apoptotic pathways in STZ-induced type I diabetes. Am J Physiol Endocrinol Metab312: E190-190E203.[Crossref]
- Brestoff JR, Artis D (2015) Immune regulation of metabolic homeostasis in health and disease. Cell 161:146-160. [Crossref]
- Koyama H, Iwakura H, Dote K, Bando M, Hosoda H, et al. (2016) Comprehensive Profiling of GPCR Expression in Ghrelin-Producing Cells. Endocrinology 157: 692-704.[Crossref]
- Ertunc ME, Hotamisligil GS (2016) Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res 57: 2099-2114.[Crossref]
- Marmugi A, Parnis J, Chen X, Carmichael L, Hardy J, et al. (2016) Sorcin Links Pancreatic β-Cell Lipotoxicity to ER Ca2+ Stores. Diabetes 65: 1009-1021.[Crossref]
- Guo R, Wu Z, Jiang J, Liu C, Wu B, et al. (2017) New mechanism of lipotoxicity in diabetic cardiomyopathy: Deficiency of Endogenous H2S Production and ER stress. Mech Ageing Dev 162: 46-52.[Crossref]
- Yano T, Shimoshige S, Miki T, Tanno M, Mochizuki A, et al. (2016) Clinical impact of myocardial mTORC1 activation in nonischemic dilated cardiomyopathy. J Mol Cell Cardiol91: 6-9.[Crossref]
- Klein BY, Tamir H, Hirschberg DL, Ludwig RJ, Glickstein SB, et al. (2016) Oxytocin opposes effects of bacterial endotoxin on ER-stress signaling in Caco2BB gut cells. BiochimBiophys Acta 1860:402-411. [Crossref]
- Xuan YL, Wang Y, Xue M, Hu HS, Cheng WJ, et al. (2015) In rats the duration of diabetes influences its impact on cardiac autonomic innervations and electrophysiology. AutonNeurosci189:31-36. [Crossref]
- Song S, Seo HH, Lee SY, Lee CY, Lee J, et al. (2015) MicroRNA-17-mediated down-regulation of apoptotic protease activating factor 1 attenuates apoptosome formation and subsequent apoptosis of cardiomyocytes. BiochemBiophys Res Commun465: 299-304.[Crossref]
- Yaser Abdallah, Sascha A Kasseckert,Wisam Iraqi,Maher Said,Tayyab Shahzad, et al. (2011) Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med 15:2478-2485. [Crossref]
- Santulli G, Xie W, Reiken SR, Marks AR (2015) Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 112:11389-11394. [Crossref]
- Chung IS, Kim JA, Choi HS, et al.(2013) Reactive oxygen species by isoflurane mediates inhibition of nuclear factor kappaB activation in lipopolysaccharide-induced acute inflammation of the lung. AnesthAnalg 116:327-335. [Crossref]
- Saotome M1, Katoh H, Yaguchi Y, Tanaka T, Urushida T, et al. (2009) Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am J Physiol Heart CircPhysiol 296:H1125-1132. [Crossref]
- Wu S, Lu Q, Wang Q, Ding Y, Ma Z, et al. (2017) Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 136: 2248-2266.[Crossref]
- L Gomez,P-A Thiebaut, M Paillard, S Ducreux,M Abrial, et al. (2016) The SR/ER-mitochondria calcium crosstalk is regulated by GSK3beta during reperfusion injury. Cell Death Differ 23:313-322. [Crossref]
- Sunaga D, Tanno M, Kuno A, Ishikawa S, Ogasawara M, et al. (2014) Accelerated recovery of mitochondrial membrane potential by GSK-3β inactivation affords cardiomyocytes protection from oxidant-induced necrosis. PLoS One 9: e112529.[Crossref]
- Li H, Murphy T, Zhang L, Huang B, Veitla V, et al. (2016) β1-Adrenergic and M2 Muscarinic Autoantibodies and Thyroid Hormone Facilitate Induction of Atrial Fibrillation in Male Rabbits. Endocrinology 157: 16-22.[Crossref]
- Yu J, Hu M, Wang Y, Zhang Q, Xu W, et al. (2018) Extraction, partial characterization and bioactivity of polysaccharides from Senecio scandens Buch.-Ham. Int J BiolMacromol109: 535-543. [Crossref]