Abstract
We used RNA sequencing (RNA-seq) to explore similarities and differences among gene co-expression in differentiated motor neurons (MN) derived either from commercial human embryonic stem cell (hESC)-derived neural stem cells (H9-NSC) or peripheral mononuclear blood cells (PMBC) reprogrammed by us to iPSC then neuralized to NSC (Ctl-NSC-MN), compared to adult motor neurons isolated with laser- capture microdissection (LCM) from cervical spinal cord sections (CTL-LCM_MN). Log2 gene expressions (8904 gene pairs) were highly correlated and very similar between H9-NSC-MN and Ctl-NSC-MN, but were less correlated with CTL-LCM_MN gene expression. Miru®, a 3-dimensional gene expression network viewer, showed that combined H9-NSC-MN and Ctl-NSC-MN gene expression profiles formed a single group with 25 individual clusters, whereas CTL-LCM-MN gene expressions formed mostly a single group with 129 clusters. There was very little gene overlap between the two groups’ clusters, which showed reduced edges (connections) among genes in the CTL-LCM_MN clusters. DAVID analysis revealed substantial differences between the two tissue sources (H9-NSC-MN/CTL-NSC-MN cells vs spinal cord CTL-LCM_MN).
Mitochondrial complexes I and V gene ontology (GO) families were observed in both populations. Our results show that at the level of transcriptomes, “young” hESC/iPSC- NSC-derived MN’s grown by themselves in 2-dimensional culture are substantially different compared to MN’s living for decades in a spinal cord matrix and attached to their physiological peripheral muscle targets. Use of such NSC-derived MN’s for therapy screening should be undertaken with caution.
Key words
neurons, embryonic stem cells, induced pluripotential stem cells, neural stem cells
Significance
One of the major limitations for understanding etiologies of and developing treatments for neurodegenerative diseases is that the affected tissue (brain, spinal cord) is not normally accessible during life. Stem cells (SC) that are pluripotential (P), derived either from human embryos (ePSC) or induced from adult human tissues (iPSC), can be converted into many cell types, including neurons. We used RNA sequencing to compare motor neurons (MN) isolated from human spinal cords (SC-MN) to motor neurons created from human ePSC’s or iPSC’s. We found that ePSC- and iPSC- derived MN’s highly resembled each other but had limited overlap with SC-MN’s.
Introduction
Development of somatic cell reprogramming technologies has allowed the creation of multiple types of differentiated cells derived from induced pluripotential stem cells (iPSC) [1]. In neuroscience, where the central nervous system is not normally accessible during life, iPSC technology holds the promise for creation of disease models during life in which vulnerable neuronal populations can be created in vitro and used to test hypotheses about disease origins and treatments [2].
In order for iPSC-derived neural cells to serve as faithful models in diseases, particularly neurodegenerative diseases where vulnerable neuronal populations slowly die over years, the iPSC-derived neurons should resemble their vulnerable adult counterparts.
On this point there is general consensus; the disagreements appear to derive mostly around the operational definition of “resemble”. To date, many investigators demonstrated development of electrophysiological similarities in iPSC-derived neurons as indicators of resemblance [3,4]. Whether this definition is appropriate awaits the identification of a molecular candidate from such iPSC-neurons that shows therapeutic activity in the adult human neurodegenerative condition. While this is admittedly a “pragmatic” definition of resemblance between iPSC-derived and adult vulnerable neurons, we propose it is a reasonable utilitarian arbiter of “resemble”.
In the case of iPSC-derived motor neurons, we and others have shown electrophysiological maturation, to the extent that current-induced depolarizations through classical Na+ channels and repolarizations through classical K+ channels occur [4,5]. Greater time in cell culture increases current-induced “spontaneous” depolarizations, suggesting greater “maturity” (O’Brien, et al, under review). What is not clear is the extent of MN differences among ePSC/iPSC-derived MN’s and in situ adult MN’s at the level of their transcriptomes, which reflect the myriad of signaling events stimulating transcription and RNA decay of the thousands of genes normally expressed in each cell.
Such an analysis can be undertaken using systems biology approaches to try and understand which gene families are affected. We carried out paired-end, high-resolution RNA sequencing on H9-NSC-derived and Ctl iPSC/NSC-derived MN’s after 21 days of MN differentiation culture, compared to populations of MN’s isolated by laser-capture microdissection (LCM) from control (CTL) adult cervical spinal cord (n=6). After bioinformatics processing and alignment against the current version of the human genome (hg38), we estimated gene expression values as FPKM (Fragments Per Kilobase of exon sequenced per Million reads). We used these values in a novel 3D network visualization software (Miru®/Markov clustering algorithm, www.kajeka.com) to seek similarities and differences among the groups of MN’s and used DAVID to define gene ontology (GO) families represented in the gene clusters created by Miru®. Our results indicate that “young” MN’s created from human embryonic stem cell-derived neural stem cells, or Ctl peripheral blood mononuclear cell-derived iPSC-neural stem cells are very similar to each other. But both are different in significant ways compared to adult MN’s isolated from cervical spinal cord sections. Our findings provide a cautionary note to the use of iPSC-derived MN’s, as presently created, for modeling adult neurodegenerative diseases that involve motor neurons, such as amyotrophic lateral sclerosis or spinal muscular atrophy. Whether such MN models can deliver effective therapies remains to be seen.
Methods
Production of MN’s from either commercially obtained H9-NSC’s or Ctl iPSC-NSC’s produced by us is described in [5]. Isolation by laser-capture microdissection of spinal motorneurons is described in [6]. Sequencing libraries were prepared from total RNA extractions/purifications as described in [7] and [6]. Paired-end RNA multiplex sequencing was carried out by Cofactor Genomics (www.cofactorgenomics.com).
Sequencing files were decompressed, examined in FastQC, then sequencing ends from minimal Phred score 20 (99% sequencing accuracy) sequences were removed using Trimmomatic. Resulting files were aligned to the hg38 human genome using Tophat2/Bowtie2, and transcript abundance was estimated using Cufflinks. Details of bioinformatic processing and scripts are provided in [7]. Presence of MN markers in LCM-isolated spinal cord MN’s is detailed in [6].
MN’s produced in culture were exposed to 0.01% DMSO as a vehicle control for a separate study [8]. RNA-seq was carried out on two independent cultures of each cell type and combined for analysis. Graphs were produced in Excel (Microsoft) and Prism (Graphpad). Miru® software was obtained from Kajeka. All computer operations were performed on Macintosh computers using OSX 10.10 or 10.11.
Results
Differentiated hESC-derived MN’s and adult iPSC-derived MN’s possess similar transcriptomes
We processed expressed genes that had FPKM values >2.0 from MN cultures after 21 days of differentiation, derived from either H9 (hESC)-NSC’s (n=9853 genes) or Ctl PBMC-iPSC-NSC’s (n=9730 genes) exposed in duplicate to DMSO vehicle as part of a drug response experiment. Both cell lines showed substantially increased expression of the motor neuron markers HB9 and ISL1 [5].
Figure 1 is a “biologically blind” X-Y plot of the log2 mean FPKM values (>2.0) for MN’s derived from Ctl iPSC-NSC compared to those derived from H9 NSC’s. A total of 8904 gene pairs could be plotted, and there is a very high correlation coefficient (r=0.92) between the log2 FPKM’s of the gene pairs. This plot demonstrates that the two MN populations are very similar to each other at the level of gene expression.
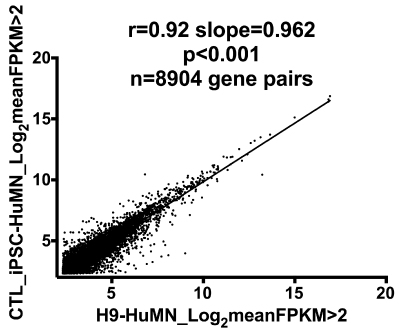
Figure 1. X-Y plot of log2 FPKM values (>2.0) of 8904 gene pairs from Ctl-iPSC-NSC-HuMN’s compared to H9-NSC-HuMN’s. The high correlation coefficient (0.92) supports great similarity between these two cell groups.
Differentiated hESC-derived MN’s and adult iPSC-derived MN’s cluster together but correlate not as well with gene expressions in CTL-LCM_MN’s
The FPKM values in CSV (comma-separated values) format (Table S2) were used as input to Miru® network visualization software (version 1.4) operating in a Mac OSX environment (v 10.10.5). Filters used included minimum Spearman correlation of 0.9, and graphs were constructed using the Fast Multipole Multilevel Method (FMMM) with graphical correlations of 0.95.
Figure 2 shows the Miru®-generated gene expression clusters for combined data from Ctl iPSC-NSC-derived MN’s and H9-NSC-derived MN’s. Markov clustering algorithm was applied and yielded the individual color-coded clusters. The CSV files used for creation of these networks are provided in Table S2. Miru® allows network identification of “clusters”, meaning genes that each form a node connected to other genes by “edges”. With this approach we found 25 individual gene clusters, of which the mean +/- S.D. of the top 12 (based on connection numbers) are plotted in Figure 3 (right group).
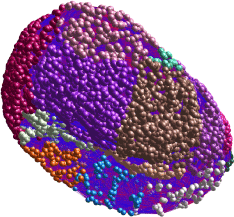
Figure 2. Miru® 3-D rendering of gene expression clusters from combination of FPKM values of H9-NSC-HuMN’s and Ctl-iPSC-NSC-HuMN’s. Markov clustering algorithm was applied, yielding the individual clusters (total n=25) that are color-coded. The mean connections among individual genes in the top 12 clusters are plotted in Figure 3.
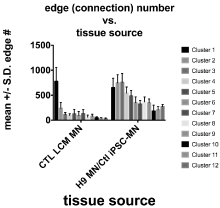
Figure 3. Mean +/- S.D. number of connections for the genes in the first 12 clusters from CTL-LCM-MN’s captured from cervical spinal cords (left group, see Figure 4), compared to those in the first 12 clusters from the combined H9-NSC-HuMN’s + Ctl-iPSC-NSC- HuMN’s (shown in Figure 2).
We carried out an identical approach for the gene expression data from the CTL LCM- isolated MN’s from six adult cervical spinal cords. Miru identified 129 clusters for this group (Figure 4); the connection numbers for the top 12 clusters are shown in Figure 3 (left group). Most of these clusters were smaller than those found in the H9-MN/CTL- iPSC-MN group (Figure 3). For Cluster 1, among the largest from each tissue type, there was a small overlap (~15-20%) between the genes identified by Miru (Figure 5) as belonging to Cluster 1. Figure 6 shows that at the gene expression level the H9-NSC-MN’s and Ctl-NSC-MN’s correlated less strongly (r=0.55, 0.50, respectively) with the gene expressions in CTL- LCM-MN’s.

Figure 4. Miru® 3-D rendering of gene expression clusters (n=129) from combination of FPKM values of the six populations of MN’s isolated by LCM from CTL cervical spinal cords. Markov clustering algorithm was applied, yielding the individual clusters that are color-coded. The mean connections among individual genes in the top 12 clusters are plotted in Figure 3.
Supplemental Table 1 compares the GO families in the clusters identified in the two different tissues (cultured MN cells compared to MN’s isolated from adult spinal cords). There was very little similarity in GO families represented, but notably both complexes I and V of the mitochondrial respiratory/OXPHOS chain were represented in both tissues.
Discussion and conclusion
New models for human nervous system diseases are greatly needed, as available animal models fail repeatedly to translate experimental therapies well into adult humans. There are multiple reasons why this might occur, and it is not the purpose of this paper to debate the utility of existing animal models to guide therapy development.
In contrast to diseases such as cancer, where the diseased tissue can frequently be studied in isolation, nervous system diseases rarely provide such access to living tissues over the course of decline. In that context interest has arisen in using reprogrammed somatic cells from disease-burdened adults, that are altered genetically into a pluripotential state, then changed to mimic one or more early neural cell phenotypes. Such induced pluripotential stem cells (iPSC’s), altered to a neural fate such as neural stem cells (NSC’s), can be propagated indefinitely and changed phenotypically into astrocytes or neurons.
Neurons derived from iPSC’s/NSC’s can possess electrophysiological properties similar to adult neurons and express phenotypic markers of neurons, including neurotransmitter synthesis, release and catabolism systems [3]. The similarities and differences among these in vitro cell models and neurons that have differentiated in situ and lived for decades in an adult CNS 3-dimensional matrix are critical for using such cell models to develop molecular therapies.
In that context, we have used RNA-seq technology to acquire gene expression data on two types of NSC’s differentiated into motor neurons, compared to endogenous motor neurons isolated by LCM from adult cervical spinal cords. The MN’s derived from commercially obtained human H9 NSC’s and PBMC-derived iPSC’s that we created from a disease-free individual, then neuralized to NSC phenotype, were very similar to each other using correlation of expression of >8900 gene pairs. However, both NSC- derived MN populations’ gene expressions were significantly less correlated with mean values of adult LCM-isolated CTL cervical spinal cord MN’s.
Miru®/Markov-based network clustering revealed that the NSC-derived MN’s expressed both many fewer and larger gene clusters and different GO families compared to the adult LCM-isolated MN’s. Notably, both groups showed significant presence of GO families from complexes I and V of the mitochondrial respiratory chain. These observations are consistent with results from earlier studies implicating mitochondrial dysfunction as a consistent component of ALS, a major neurodegeneration of spinal motor neurons. [6,9-11]
Both cultured cell pop2021 Copyright OAT. All rights reservthat they have existed in MN differentiation media for only 21 days, and immunohistochemical data indicate that only a fraction of cells obtained appear to express a full MN phenotype. In spite of this limitation, the gene expression differences between the H9/CTL NSC and LCM-isolated MN’s are substantial and suggest that these cell types bear limited resemblance to each other.
This “degree of separation” is visualized in Figure 5, where only ~15-25% of expressed genes in Cluster 1 (one of the largest clusters in both groups) overlapped. For Cluster 2, a comparable figure for shared genes is an overlap of 4.2% (H9/Ctl NSC-MN) and 11.3% (CTL-LCM-MN). Our findings suggest that based on their gene expression differences, use of NSC- derived MN’s to screen potentially therapeutic drugs for MN disorders requires caution. We note that we did not use NSC-derived MN’s from subjects with a particular MN disease, to compare with LCM-isolated MN’s from spinal cords derived from subjects who died with the same disease (an example would be amyotrophic lateral sclerosis, ALS). We hope to be able to examine this situation in a future study.
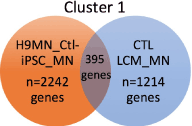
Figure 5. Venn diagram showing overlap in genes identified in cluster 1 of H9-MN’s/Ctl-MN’s compared to CTL-LCM-MN’s captured from cervical spinal cords. Most of the genes in both populations did not overlap.
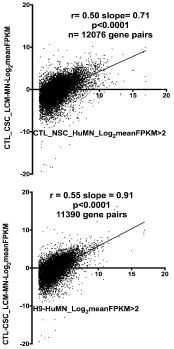
Figure 6. Reduced correlations of gene expression in Ctl-MN’s (top) or H9-MN’s (bottom) compared to that found in CTL-LCM-MN’s isolated from human cervical spinal cords.
Our study has several important limitations, a few of which are listed below:
First, we used only one gene expression clustering algorithm (Miru®, followed by Markov clustering). Miru® has the advantage of network visualization in a 3-D environment and detailed analysis of individual clusters found by the Markov algorithm. We utilized this feature to discern both cluster size (ie, number of connections among genes in each cluster) and DAVID GO families represented by each cluster. These features contributed to our finding multiple differences between the two populations of MN’s (derived from H9/Ctl-NSC’s compared to LCM-isolated from adult cervical spinal cords).
Second, we were comparing MN cells in 2D culture, not connected to any targets, certainly not physiological targets (i.e., muscle endplates), to intact MN’s in cervical spinal cords that presumably had been connected to physiological targets for decades. That the two different tissues had substantially different gene expression profiles is therefore not surprising. We hope to discern whether the availability of physiological targets (ie muscle cells in culture) changes the gene expression profiles of the in vitro MN’s.
Third, our study of LCM-isolated MN’s used population averages for gene expression. We typically collected hundreds-thousands of individual MN’s from each spinal cord sample, and current RNA-seq technology does not allow sequencing of single MN’s. Even with population averaging across LCM-MN populations, we observed variations that suggest substantial heterogeneity within this group.
Fourth, we did not enrich for MN phenotypes (such as with FACS) in our ePSC/iPSC-NSC cultures differentiated for 21 days. Thus, we cannot assume that all NSC’s were converted to MN’s.
In summary, we report that MN’s derived from neuralized NSC’s from both H9 hESC’s and Ctl iPSC’s, transformed to MN phenotypes in culture, bear limited molecular genetic relationship to MN’s isolated by LCM from CTL adult cervical spinal cord sections. (A notable exception to this observation involves presence of GO families for mitochondrial complexes I and V in both tissues.) This discrepancy should be considered before this cell population is used for therapy screening. Improvements may result from the following: 1. Use of disease-bearing subjects for PBMC-iPSC-NSC-MN generation compared to MN’s isolated from spinal cord sections of those who died with an identical condition; 2. Provision of physiological targets for MN’s in culture; 3. “aging” of the MN’s in culture.
Acknowledgement
This research was generously supported by ALS Worldwide, the VCU Parkinson’s Research Center, through the MCV Foundation, and Neurodegeneration Therapeutics, Inc.
Conflicts of interest
None of the authors has any conflicts of interest with this research.
Author contributions
All authors participated in various aspects of experimental design. DGB did RNA extractions, purifications, helped with DMSO exposures of MN’s, and made/quantitated all RNA sequencing libraries; ACL captured all MN’s from cervical spinal cord sections and carried out RNA extractions and purifications; LCO’B developed the protocol for MN differentiation of NSC’s and carried out all MN differentiation experiments and DMSO exposures. JPB carried out bioinformatics and Miru® analyses, and wrote the manuscript draft. All authors have seen and approved the final manuscript.
References
- Ho PJ, Yen ML, Yet SF, Yen BL (2012) Current applications of human pluripotent stem cells: possibilities and challenges. Cell Transplant 21: 801-814. [Crossref]
- Glavaski-Joksimovic A, Bohn MC (2013) Mesenchymal stem cells and neuroregeneration in Parkinson's disease. Exp Neurol 247: 25-38. [Crossref]
- Grow DA, Simmons DV, Gomez JA, Wanat MJ, McCarrey JR, et al. (2016) Differentiation and Characterization of Dopaminergic Neurons From Baboon Induced Pluripotent Stem Cells. Stem Cells Transl Med 5:1133-1144. [Crossref]
- Burkhardt MF, Martinez FJ, Wright S (2013) A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 56: 355-364.
- O'Brien LC, Keeney PM, Bennett JP, Jr (2015) Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev 24: 1984-1994. [Crossref]
- Ladd AC, Brohawn DG, Thomas RR, Keeney PM, Berr SS, et al. (2017) RNA-seq analyses reveal that cervical spinal cords and anterior motor neurons from amyotrophic lateral sclerosis subjects show reduced expression of mitochondrial DNA-encoded respiratory genes, and rhTFAM may correct this respiratory deficiency. Brain Res 1667: 74-83. [Crossref]
- Brohawn DG, O'Brien LC, Bennett JP, Jr (2016) RNAseq Analyses Identify Tumor Necrosis Factor-Mediated Inflammation as a Major Abnormality in ALS Spinal Cord. PLoS One 11: e0160520. [Crossref]
- Bennett JP, Jr., O'Brien LC, Brohawn DG (2016) Pharmacological properties of microneurotrophin drugs developed for treatment of amyotrophic lateral sclerosis. Biochem Pharmacol 117:68-77. [Crossref]
- Ladd AC, Keeney PM, Govind MM, Bennett JP Jr (2014) Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromolecular Med 16: 714-726. [Crossref]
- Rice AC, Keeney PM, Algarzae NK, Ladd AC, Thomas RR, et al. (2014) Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer's disease hippocampi. J Alzheimer's Dis 40:319-330. [Crossref]
- Sasaki S, Iwata M (2007) Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66: 10-16. [Crossref]