Abstract
Historically, forensic STR panels have been unsuccessful for population assignment due to the limited ancestry information that can be derived from the non-coding STR loci and the low number of loci included in the panel. However, given the recent adoption of expanded (16+ loci) and ‘mega-plex’ (23+ loci) STR panels, the ability to identify source population groups may be improved. This study assessed the impact of increasing locus number on population assignment under different analysis conditions using a published US population dataset comprised of individuals from the African American, Caucasian, Hispanic and Asian populations. The Bayesian clustering programme STRUCTURE was used to assess first, whether increasing the number of loci and the inclusion of known sample population data enabled greater resolution between the four populations in the dataset, and second, the utility for population assignment using criteria based on inferred ancestry scores. Results suggest that increasing the number of loci and including population of origin data allowed the identification of more distinct populations, with three primary populations being observed; African American, Asian, and Caucasian/Hispanic. The close grouping of the Caucasian and Hispanic populations is supported by their recently common ancestry from Western Europe. The ability of the programme to support population assignment to each of the four existing populations was assessed through the application of population and panel specific assignment thresholds based on the inferred ancestry scores obtained from the analysis programme. Predictive accuracy based on a training dataset of 984 individuals suggest that assignment accuracy is > 96% across the four populations and can reach 100% under some test conditions. The accuracy was > 90% when blind testing was performed on 40 ‘unknown’ individuals. As such, the approach described is considered within the acceptable range for a presumptive test and can be performed using data already collected as part of routine forensic investigations.
key words
population assignment, structure, forensic str, geographic origin, evidential weight
Introduction
Inference of the ethnic origin of a suspect from their DNA recovered from a crime scene sample can act as ‘investigative intelligence’ and help enforcement agencies concentrate their resources in the absence of any other suspect specific information. This idea is not new and has been explored extensively in the literature through the development and application of assignment approaches that use genetic markers to identify unique genetic groups or populations [1-3]. The specific identification of Ancestry Informative Markers (AIMs) panels that are particularly powerful at inferring ethnic origin has also been the focus of much research and the assessment of different classes of molecular marker has slowly moved from mitochondrial DNA (mtDNA) sequence variation, through autosomal and Y linked Short Tandem Repeat (STRs) markers to Single Nucleotide Polymorphisms (SNPs) [4-8]. SNP markers are currently considered the most applicable for use due to deficiencies displayed by other markers; uniparental markers (mtDNA and Y-STRs) typically require large datasets to be useful while autosomal STRs are not considered informative by some researchers in numbers less than 50 [9].
A number of biogeographically informative SNP marker panels have been developed [9,10], and with the adoption of Massively Parallel Sequencing (MPS) as a resource in forensic genetics, commercial MPS forensic panels are available that amplify both traditional STR, Y-STR phenotypic and AIM markers in a single reaction run [11-13]. However, despite the promise of MPS and the development of AIM panels, the forensic genetics community continues to use traditional multiplex-PCR kits for the amplification and size separation of STRs through capillary electrophoresis (CE). Reasons for the slow adoption of MPS technologies include high per sample cost, increased processing time and uncertainty around data handling and ethics [13,14]. As such, laboratories continue to use CE approaches and have seen the number of loci included in commercial panels virtually double in the last five years. For example, the European standard set (ESS) now comprises 17 STR loci and the American CODIS system comprises 20 core loci [15-18], while both the commercially available GloablFiler and PowerPlex Fusion kits boast an impressive 22+ STR loci [19,20]. Consequently, the increasing number of STR loci may now enable better resolution between populations, thus making expanded and mega-plex STR kits suitable for genetic differentiation between populations [21] and the inference of ethnic origin. Such an approach would be useful if labs continue to use CE methods for the foreseeable future.
Population assignment requires the use of a mathematical model that groups an unknown individual to a putative population and can be used to detect dispersal, hybridization, genetic mixture, origin of specific individuals, population delineation and structure [22]. Common population assignment models include Bayesian assignment, frequency-based, and Bayesian clustering approaches. The Bayesian assignment approach developed by Rannala and Mountain [23] calculates the posterior probabilities that a genotype is observed at a locus when the individual belongs to each putative population. The probability is then determined for each locus (assuming no linkage) and multiplied, and results are provided as the posterior probability with lower values indicating rarer events. This approach has been used in the detection of poaching hot-spots [24], differentiation between closely related species [25] and the identification of illegally translocated deer [26]. An alternative, frequency-based method developed by Paetkau et al., [27] calculates genotype likelihood ratios and determines the probability that the genotype groups with each population using Monte Carlo resampling. This approach has been used to assign individual dogs to their population of origin [28], identify livestock predators [29], and to detect fishing competition fraud [30]. These two approaches, popular in molecular ecology, have seen little application in human population assignment, where research has concentrated on the development of bespoke models [7,10,31]. Perhaps one of the most common approaches to investigate human population genetic differentiation is the Bayesian clustering method developed by Pritchard et al., [32] which uses multi-locus genotype data to infer the number of distinct genetic clusters (populations) based on the allele frequencies observed in each population. Individuals across the dataset are assigned to single populations, or to multiple populations if admixture is detected. This approach has been successfully used to map clines in human population genetic structure with geography [33-35].
This proof of concept research aims to establish whether the increased number of loci used in expanded and mega-plex STR panels improves assignment accuracy and asks whether there is scope for a ‘presumptive’ population assignment test for forensic laboratories continuing to use CE based systems.
Methods
Sample data
STR profiles from 1036 individuals previously reported in ref. [36,37] were downloaded from Promega (https://www.promega.com/products/pm/genetic-identity/population-statistics/allele-frequencies/). Samples with genotyping errors as highlighted by Steffen et al., [38] were removed from the analysis resulting in a final sample set of 1024 individuals. This final dataset represents four common American population groups; African American (AA; n = 338), Caucasian (Ca; n = 358), Hispanic (His; n = 232) and Asian (As; n = 96). Genotype data were reformatted with new allele nomenclature (see supplemental Table 1) to allow for software analysis. Y-linked markers were removed from the dataset due to software input criteria, resulting in a panel comprising 30 autosomal STR loci. Separate input files were created to represent commonly used commercial STR profiling kits, each with a different number of STR loci; PowerPlex 16 (16 autosomal loci), AmpFlSTR NGM SElect (17 autosomal loci), AmpFlSTR GlobalFiler™ (22 autosomal loci, and excluding the DYS391 Y-linked locus) and PowerPlex Fusion (23 autosomal loci, and excluding the YINDEL Y-linked locus).
Table 1: Parameter Settings and estimated K for three different analyses using STRUCTURE.
Analysis
Parameter |
Purpose |
|
STRUCTURE Software Conditions |
|
Estimated K using 3 methods |
Run
Conditions |
Population
Model |
LOCPRIOR |
POPFLAG |
STR Panel and Locus number (n) |
Hightest InPD |
ΔK |
Plateau InPD |
1 |
Structure
Identification |
100k burnin,
100k MCMC
reps |
Admixed |
No |
No |
PowerPlex (16)
NGM Select (17)
Global Filer (22)
PowerPlex Fusion (23)
Combined Panel (30)
|
3
3
3
4
5 |
2
2
2
2
2 |
2
2
3
3
4 |
2 |
Structure
Identification |
100k burnin.
100k MCMC
reps |
Admixed |
yes |
No |
PowerPlex (16)
NGM Select (17)
Global Filer (22)
PowerPlex Fusion(23)
Combined Panel (30)
|
3
4
5
4
4 |
2
2
2
2
2 |
2
3
3
3
3 |
3 |
Setting Population Assignment Criteria and Assessing |
100k burnin,
100k MCMC
reps |
Admixed |
yes |
yes |
PowerPlex (16)
NGM Select (17)
Global Filer (22)
PowerPlex Fusion(23)
Combined Panel (30)
|
5
5
5
5
5 |
2
2
2
2
3 |
3
3
3
3
3 |
Population structuring
The Bayesian clustering method STRUCTURE [32] was first used to identify the likely number of distinct genetic clusters (populations; K) existing in the data for each of the STR profiling kits. Two different analysis parameters (1 and 2) were initially tested to explore population structuring with and without the inclusion of known sample population data (Table 1). Each parameter set underwent five analysis iterations at each possible K (1-5). The optimal K was identified using three different approaches, avoiding the use of a single ad-hoc approach [32,39]: first, the highest mean log-likelihood value (lnPD) method outlined in ref. [32] was used; second, the ΔK method detailed in ref. [40] was calculated using the web-based STRUCTURE HARVESTER programme [41]; and third, the point at which the lnPD values begin to plateau as outlined in ref. [33]. CLUMPAK [42] was used to visualise the data. The use of the LOCPRIOR setting in parameter set two was shown to identify fine scale population differences more effectively and was selected for use when assessing population assignment.
Population assignment
Assignment accuracy was assessed under the expected number of populations (K=4) using analysis parameter three (Table 1) for each of the five STR profiling kits under study. This parameter set included the use of the POPFLAG feature in STRUCTURE that allows the assignment of individuals of unknown origin to a dataset containing individuals of known origin. Ten random individuals from each population were labelled as ‘unknowns’ resulting in a total of 984 known and 40 ‘unknown’ samples run using analysis parameter set three. Predicted assignment accuracy was calculated based on the reduced data set of 984 individuals, which was used as a ‘training’ sample set to develop suitable acceptance criteria for assignment. To do this, the mean inferred ancestry scores for each individual from the five iterations when K=4 were calculated in CLUMPAK and the distributions for each population in each of the four clusters plotted. Assignment criteria for each population for each STR kit were then determined by setting a threshold for inferred ancestry score for each of the four genetic clusters. This approach is analogous to the setting of an analytical threshold to differentiate signal (true contribution) and noise (false contribution). Across the training dataset of 984 samples the number of individuals that were assigned to a single group using the defined criteria was calculated. An individual that satisfied all four criteria was given a score of four and categorised as either True Positives (TP) or False Positives (TP), while individuals that were given a scores of less than four were classified as True Negatives or False Negatives following the definitions supplied in Supplemental Table 2. Once categorised, these values were used to determine the predicted test sensitivity and specificity following the binary classification system outlined in ref. [43]. The ten randomly selected ‘unknown’ individuals from each population were then assessed to see how well they were assigned to the populations based on the defined criteria.
Table 2: Predicted accuracy of population assignment test for different STR panels.
Population |
Accuracy |
PowerPlex (16) |
NGMSelect
(17) |
Global Filer
(22) |
Power Plex
Fusion (23) |
Combined
Panel (30) |
African
American |
TP |
328 |
328 |
328 |
328 |
328 |
TN |
656 |
656 |
656 |
656 |
656 |
FP |
0 |
0 |
0 |
0 |
0 |
FN |
0 |
0 |
0 |
0 |
0 |
Sensitivity |
100% |
100% |
100% |
100% |
100% |
Specificity |
100% |
100% |
100% |
100% |
100% |
Caucasian |
TP |
348 |
348 |
348 |
348 |
348 |
TN |
636 |
636 |
632 |
631 |
635 |
FP |
0 |
0 |
4 |
5 |
1 |
FN |
0 |
0 |
0 |
0 |
0 |
Sensitivity |
100% |
100% |
100% |
100% |
100% |
2021 Copyright OAT. All rights reserv
Specificity |
100% |
100% |
99.3% |
99% |
99.8% |
Hispanic |
TP |
222 |
222 |
222 |
222 |
222 |
TN |
676 |
762 |
728 |
738 |
760 |
FP |
0 |
0 |
34 |
24 |
2 |
FN |
0 |
0 |
0 |
0 |
0 |
Sensitivity |
100% |
100% |
100% |
100% |
100% |
Specificity |
100% |
100% |
96% |
97% |
99.7% |
Asian |
TP |
86 |
86 |
86 |
86 |
85 |
TN |
898 |
898 |
898 |
898 |
898 |
FP |
0 |
0 |
0 |
0 |
0 |
FN |
0 |
0 |
0 |
0 |
1 |
Sensitivity |
100% |
100% |
100% |
100% |
98.8% |
Specificity |
100% |
100% |
100% |
100% |
100% |
|
TP: True Positives, TN: True Negatives, FP: False Postives, FN: False Negatives. Sensitivity =TP/(TP4FN) and Specificity = TN/(TN4FP) calculated according to Ref. [43].
Results and Discussion
The STRUCTURE analysis shows that the number of distinct genetic groups (K) identified varies depending on which method is used in the estimation (Table 1). The recommended method for interpreting the correct K using STRUCTURE is to not use a single add-hoc approach as they each have limitations [32,39]. The ΔK method outlined in ref. [40] is predicted to underestimate the number of distinct clusters, while taking the highest mean log-likelihood value (lnPD) is thought to overestimate. Using the plateau approach [32] generally identifies a K value between the two methods and was considered the most appropriate in this study. Using this method the number of clusters identified increases with locus number under STRUCTURE analysis parameter 1 with two distinct clusters identified for both PowerPlex 16 and AmpFlSTR NGM Select (16 and 17 loci respectively), three clusters for GlobalFiler and PowerPlex Fusion (22 and 23 loci respectively), and four clusters for the combined panel of 30 loci. This is seen to a lesser extent in the results for parameter 2, with two distinct clusters identified for PowerPlex 16 and three clusters for the other four marker panels. Highest lnPD followed a similar trend of increasing K with number of loci and provided the greatest estimates of K, while ΔK showed little change across all analyses. An increase in the ability to identify more clusters with more loci was expected as it has previously been observed that using fewer markers reduces STRUCTURE’s ability to cluster into a higher number of populations as less genetic variation is observed [21,44]. Previous research has shown that the ability to distinguish between populations is improved with more markers but also as a function of how informative the loci are [40] and the presence of rare or private alleles [45].
There are slight differences in the returned K value for analysis parameter sets 1 and 2, with a higher K obtained with less loci for parameter set 2 using both the plateau and highest lnPD. Parameter set 2 used the LOCPRIOR setting in STRUCTURE which allows the software to use information associated with the samples such as phenotype, in this instance the sample population of African American, Caucasian, Hispanic or Asian, to support the resolution of fine scale clusters [32]. Analysis under these parameters provided greater resolution to the inferred ancestry scores leading to more confidence in the population clusters (Figure 1), while not having a substantial impact on the number of clusters observed. For application in a forensic setting, the samples can be considered as a database containing samples of known origin allowing the use of the LOCPRIOR setting for population assignment.
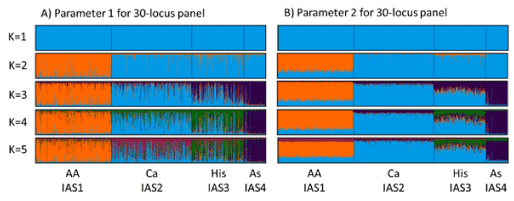
Figure 1. Stacked bar graphs for 1024 individuals for 30-locus panel showing inferred ancestry scores for population genetic clusters identified by different colours. IAS1 = Orange; IAS2 = Light Blue; IAS3 = Green; IAS4 = Purple; IAS5 = Pink. Population sample data grouped showing African American (AA), Caucasian (Ca), Hispanic (His) and Asian (As). Parameter 2 shows better differentiation between the genetic clusters and was used for setting threshold criteria for population assignment.
Genetic differentiation begins to be observed when K=2 with the African American population showing a distinct cluster while all other populations group as a single cluster (Figure 1). The next cluster to appear when K=3 is the separation of the Asian population from the Caucasian and Hispanic populations. This pattern is expected as human populations have their geographic origins in Africa, with dispersal first east through the Asian continent and then again later west through the European continent [46]. In addition, both the Asian and European populations are thought to have undergone population bottlenecks in their life histories [47] that will have led to variation in their allele frequencies. The grouping of Caucasian and Hispanic populations when K=3 can also be explained due to the admixed nature of the Hispanic population in America, which is derived from the influx of Europeans into the native population, and so shares recent common ancestry with the Caucasian population [48]. Differences between the Hispanic and Caucasian populations begin to emerge when K=4 and 5 and is apparent for the 30-locus panel shown in figure 1.
Calculating the predictive accuracy of population assignment was performed on the 984 individuals analysed under parameter 3 using data for K=4 to enable assignment to the four known populations. While the STRUCTURE results provide most support for three clusters (although K=5 for highest lnPD), the distribution of inferred ancestry scores for the 30-locus panel shows there is some clear pattern of differentiation between the four populations over four clusters (Figure 2). Threshold values, as shown for the African American population in Figure 2, were determined from these distributions and formed the four criteria for assignment to a population.
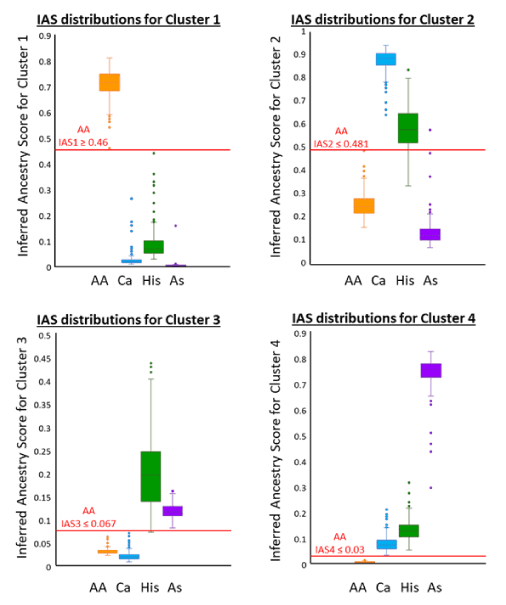
Figure 2. Box and Whisker plots showing distribution of Inferred Ancestry Scores (IAS) to each of the four genetic clusters identified by STRUCTURE for the 30-locus panel. Where there is overlap in one distribution there is usually clear separation at another which allows the setting of a contingent threshold for each population for each cluster. The thresholds and examples values for the AA population are shown in red.
Thresholds set for each population for each kit were used to assess the predicative accuracy of the assignment test. Individuals satisfying all criteria were considered positive (true or false) and any single criteria not satisfied led to the individual being considered negative (true or false). The sensitivity and specificity for the test was calculated based on the number of true/false positive and true/false negative assignments across the 984 individuals (Table 2). For the 30-locus data set the thresholding mechanism described was able to correctly assign 99% of all individuals to the correct population. A high degree of accuracy (>96%) was also shown for the two commercial mega-plex panels and those with expanded core loci, suggesting that the approach detailed here is robust and repeatable across panels, with only small fluctuations in accuracy.
Using the same thresholds and assignment criteria described above, the 40 ‘unknown’ individuals removed from the ‘training’ dataset were assigned to each of the populations (Figure 3). The sensitivity of the assignment was > 90% for all the STR panels tested and with only 8 false negatives observed; three for 30-locus panel, 1 for PowerPlex Fusion, 1 for GlobalFiler, 2 for NGM Select, 1 for PowerPlex 16. Specificity was greater than 99% with only 2 false positives observed; 1 for PowerPlex Fusion, 1 for GlobalFiler. These observed levels align closely with the predicted values based on the training data. The few samples that were not correctly assigned were outliers that did not cluster with the inferred ancestry score distribution and so were failed based on the thresholds set. Although it is likely that such outliers will continue to be observed in the wider population, the training dataset of 984 individuals is considered relatively robust and representative, and the thresholds can be refined to improve accuracy. It is important to consider that the accuracy is only based on the four test populations. The addition of further populations of interest is likely to reduce the accuracy as population genetic clusters become harder to distinguish, resulting in a wider distribution of the inferred ancestry scores. Furthermore, any individual from a population not represented in the dataset would still group with one of the populations in the absence of any representative of their own population. This is a common limitation to population assignment tests as they tend to assume all source populations are represented in the database. As such it is the authors opinion that this data be viewed as a test case for assignment rather than a usable approach for the identification of African American, Caucasian, Hispanic and Asian American individuals. Only with the addition of more population groups will the utility of the approach be truly understood. However, the high accuracy achieved would suggest there is merit in exploring the described application in more detail.
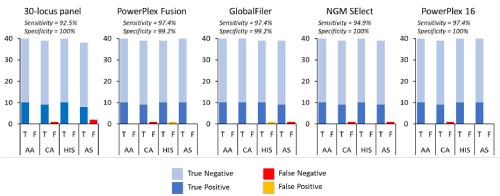
Figure 3. Accuracy of five different STR panels when assigning unknown samples to each population (AA, CA, HIS, AS). T = True assignment, F = False assignment. Sensitivity and Specificity measures provided based on average across all four populations.
The utility of a presumptive population assignment test such as that described here is important to consider in relation to other approaches. While there are better characterised panels of ancestry informative markers available, they have not all been commercialised into a quality-controlled product and are not used as part of routine analysis by most forensic laboratories. Additionally, the adoption of MPS as an approach is still some years away for many laboratories. It therefore seems prudent to understand whether there is any further information that can be derived from the use of existing STR panels. It is also important to consider that enforcement agencies already attempt to infer ethnic origin using less reliable methods than that described here. Questions of suspect skin colour and race are commonly addressed through witness statements and CCTV, both with low accuracy and subject to conscious and unconscious bias [49-51]. For example, individuals providing eyewitness testimony’s can adhere to a phenomenon known as ‘own race/ethnicity bias’ which results in a larger percentage of correct identification of race when the individual is of the same race [52]. The first study to assess the accuracy of cross-racial identification between ‘Anglo’ (non-Hispanic white), ‘black’, and Mexican American individuals found that the correct ethnicity was assigned only 44.2% of the time [53]. The results of this ethnicity bias study underlines why eye witness testimonies are not viewed by jurors as highly reliable evidence [54], but also recognises there is a balance to be struck between accuracy and evidential strength. Novel genetic assays and tools developed for forensic science are typically expected to be extremely accurate, an expectation derived from the high discriminatory power observed when performing DNA profiling. However, the development and adoption of presumptive DNA tests for ‘rapid-intelligence’ [55,56] suggest that the reality is likely to be more nuanced. As such, the authors feel that there may be some merit to the consideration of a ‘presumptive’ population assignment approach similar to that described here. Other non-forensic applications of this approach may include the inference of ancestry for supporting biogeographic research [1]. Given that STR data for human populations are routinely collected and published online it seems likely that the approach described here can be initially used to support wider research into human life history.
Summary
This preliminary study demonstrates that population assignment is possible using expanded and mega-plex STR panel with >90% accuracy. This level of accuracy is in the range of a presumptive test and the analysis can be performed using existing STR data collected as part of routine criminal casework. The use of presumptive tests to inform investigative leads is common in forensic laboratories and enforcement authorities understand the limitations of such tests. As such it is considered that this approach may provide some useful insight into the ethnic origin of unknown individuals based on their crime scene profile. Before adoption, further research should look to include a greater range of population data to understand the true utility of this approach and to assess the reproducibility of the approach. To facilitate this, the authors have included the relevant allele coding in the supplemental material for other interested groups to perform their own analysis. Once optimised the approach can be compared to other existing population assignment methods described in the literature to compare performance.
Acknowledgements
The authors would like to thank the reviewers whose advice has helped shaped this version of the manuscript. Further thanks go to Stephanie Johnson, Candice Carabini and Robert Measor who were involved in the evolution of this paper.
References
- Elhaik E, Tatarinova T, Chebotarev D, Piras IS, Calò CM, et al. (2014) Geographic population structure analysis of worldwide human populations infers their biogeographical origins. Nat Commun 29: 3513. [Crossref]
- Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, et al. (2002) Genetic structure of human populations. Science 20: 2381-2385. [Crossref]
- Ekins JE, Ekins JB, Layton L, Hutchison LA, Myres NM, et al. (2006) Inference of ancestry: Constructing hierarchical reference populations and assigning unknown individuals. Hum genomics 2: 212. [Crossref]
- Hwa HL, Lin CP, Huang TY, Kuo PH, Hsieh WH, et al. (2017) A panel of 130 autosomal single-nucleotide polymorphisms for ancestry assignment in five Asian populations and in Caucasians. Forensic Sci Med Pathol 13: 177-187. [Crossref]
- Egeland T, Bøvelstad HM, Storvik GO, Salas A (2004) Inferring the most likely geographical origin of mtDNA sequence profiles. Ann Hum Genet 68: 461-471. [Crossref]
- Jorde LB, Watkins WS, Bamshad MJ, Dixon ME, Ricker CE, et al. (2000) The distribution of human genetic diversity: a comparison of mitochondrial, autosomal, and Y-chromosome data. Am J Hum Genet 66: 979-988. [Crossref]
- Lowe AL, Urquhart A, Foreman LA, Evett IW (2001) Inferring ethnic origin by means of an STR profile. Forensic Sci Int 119: 17-22. [Crossref]
- Rosenberg NA, Li LM, Ward R, Pritchard JK (2003) Informativeness of genetic markers for inference of ancestry. Am J Hum Genet 73: 1402-1422. [Crossref]
- Phillips C, Salas A, Sanchez JJ, Fondevila M, Gomez-Tato A, et al. (2007) Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs. Forensic Sci Int Genet 1: 273-280. [Crossref]
- Zeng X, Warshauer DH, King JL, Churchill JD, Chakraborty R, et al. (2016) Empirical testing of a 23-AIMs panel of SNPs for ancestry evaluations in four major US populations. Int J Legal Med 130: 891-896. [Crossref]
- Zeng X, King JL, Stoljarova M, Warshauer DH, LaRue BL, et al. (2015) High sensitivity multiplex short tandem repeat loci analyses with massively parallel sequencing. Forensic Sci Int Genet 16: 38-47. [Crossref]
- Xavier C, Parson W (2017) Evaluation of the Illumina ForenSeq™ DNA Signature Prep Kit–MPS forensic application for the MiSeq FGx™ benchtop sequencer. Forensic Sci Int Genet 28: 188-194. [Crossref]
- Scudder N, McNevin D, Kelty SF, Walsh SJ, Robertson J (2017) Massively parallel sequencing and the emergence of forensic genomics: Defining the policy and legal issues for law enforcement. Sci Justice. 58: 153-158. [Crossref]
- Butler JM (2015) The future of forensic DNA analysis. Philos Trans R Soc Lond B Biol Sci 370. [Crossref]
- Cotton EA, Allsop RF, Guest JL, Frazier RR, Koumi P, et al. (2000) Validation of the AMPFlSTR® SGM Plus™ system for use in forensic casework. Forensic Sci Int 112: 151-161. [Crossref]
- Hares DR (2012) Expanding the CODIS core loci in the United States. Forensic Sci Int Genet 6: e52-4.
- Green RL, Lagacé RE, Oldroyd NJ, Hennessy LK, Mulero JJ (2013) Developmental validation of the AmpFlSTR® NGM SElect™ PCR Amplification Kit: a next-generation STR multiplex with the SE33 locus. Forensic Sci Int Genet 7: 41-51. [Crossref]
- Tucker VC, Hopwood AJ, Sprecher CJ, McLaren RS, Rabbach DR, et al. (2011) Developmental validation of the PowerPlex® ESI 16 and PowerPlex® ESI 17 Systems: STR multiplexes for the new European standard. Forensic Sci Int Genet 5: 436-448.
- Hennessy LK, Mehendale N, Chear K, Jovanovich S, Williams S, et al. (2014) Developmental validation of the GlobalFiler® express kit, a 24-marker STR assay, on the RapidHIT® System. Forensic Sci Int Genet 13: 247-258. [Crossref]
- Oostdik K, Lenz K, Nye J, Schelling K, Yet D, et al. (2014) Developmental validation of the PowerPlex® Fusion System for analysis of casework and reference samples: a 24-locus multiplex for new database standards. Forensic Sci Int Genet 12: 69-76. [Crossref]
- McCulloh KL, Ng J, Oldt RF, Weise JA, Viray J, et al. (2016) The genetic structure of native Americans in North America based on the Globalfiler® STRs. Leg Med 23: 49-54. [Crossref]
- Manel S, Gaggiotti OE, Waples RS (2005) Assignment methods: matching biological questions with appropriate techniques. Trends Ecol Evol 20: 136-142. [Crossref]
- Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94: 9197-9221. [Crossref]
- Manel S, Berthier P, Luikart G (2002) Detecting wildlife poaching: identifying the origin of individuals with Bayesian assignment tests and multilocus genotypes. Conserv biol 16: 650-659.
- Sorenson L, McDowell JR, Knott T, Graves JE (2013) Assignment test method using hypervariable markers for blue marlin (Makaira nigricans) stock identification. Conserv Genet Res 5: 293-297.
- Frantz AC, Pourtois JT, Heuertz M, Schley L, Flamand MC, et al. (2006) Genetic structure and assignment tests demonstrate illegal translocation of red deer (Cervus elaphus) into a continuous population. Molecular Ecology. 15: 3191-3203. [Crossref]
- Paetkau D, Calvert W, Stirling I, Strobeck C (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol 4: 347-354. [Crossref]
- Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, et al. (2004) Genetic structure of the purebred domestic dog. Science 304: 1160-1164. [Crossref]
- Caniglia R, Fabbri E, Mastrogiuseppe L, Randi E (2013) Who is who? Identification of livestock predators using forensic genetic approaches. Forensic Sci Int Genet 7: 397-404. [Crossref]
- Primmer CR, Koskinen MT, Piironen J (2000) The one that did not get away: individual assignment using microsatellite data detects a case of fishing competition fraud. Proc Biol Sci 267: 1699-1704. [Crossref]
- Graydon M, Cholette F, Ng LK (2009) Inferring ethnicity using 15 autosomal STR loci—Comparisons among populations of similar and distinctly different physical traits. Forensic Sci Int Genet 3: 251-254. [Crossref]
- Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945-959. [Crossref]
- Bamshad MJ, Wooding S, Watkins WS, Ostler CT, Batzer MA, et al. (2003) Human population genetic structure and inference of group membership. Am J Hum Genet 72: 578-89. [Crossref]
- Sun QF, Jiang L, Liu J, Zhao L, Ji AQ, Li CX (2017) Validation analysis of a 27-plex SNP panel for ancestry inference. Forensic Sci Int Genet Supple Ser 6: e603-5.
- Elhaik E, Tatarinova T, Chebotarev D, Piras IS, Calò CM, et al. (2014) Geographic population structure analysis of worldwide human populations infers their biogeographical origins. Natu commun 5: 3513. [Crossref]
- Hill CR, Duewer DL, Kline MC, Coble MD, Butler JM (2013) US population data for 29 autosomal STR loci. Forensic Sci Int Genet 7: e82-e83. [Crossref]
- Butler JM, Hill CR, Coble MD (2012) Variability of new STR loci and kits in U.S. population groups. Profiles in DNA. Available at http://www.promega.com/resources/articles/profiles-in-dna/2012/variability-of-new-str-loci-and-kits-in-us-population-groups/.
- Steffen CR, Coble MD, Gettings KB, Vallone PM (2017) Corrigendum to 'U.S. Population Data for 29 Autosomal STR Loci'. Forensic Sci Int Genet 31: e36-36e40. [Crossref]
- Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, et al. (2017) The K= 2 conundrum. Mol Ecol
- Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611-2620. [Crossref]
- Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation genetics resources 4: 359-361.
- Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res 15: 1179-1191.
- Altman DG, Bland JM (1994) Diagnostic tests. 1: Sensitivity and specificity. BMJ 308: 1552. [Crossref]
- Silva NM, Pereira L, Poloni ES, Currat M (2012) Human neutral genetic variation and forensic STR data. PLoS One 7: e49666. [Crossref]
- Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39: 53-65. [Crossref]
- Mellars P (2006) Why did modern human populations disperse from Africa ca. 60,000 years ago? A new model. Proc Natl Acad Sci U S A 103: 9381-9386.
- Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475: 493-496. [Crossref]
- Bryc K, Velez C, Karafet T, Moreno-Estrada A, Reynolds A, et al. (2010) Genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc Natl Acad Sci U S A 11: 8954-8961. [Crossref]
- Meissner CA, Brigham JC (2001) Thirty years of investigating the own-race bias in memory for faces: A meta-analytic review. Psychol Pub Poli and Law 7: 3-35.
- Wells GL, Olson EA (2001) The other-race effect in eyewitness identification: What do we do about it? Psychol Pub Poli and Law 7: 230-802.
- Brigham JC, Maass A, Snyder LD, Spaulding K (1982) Accuracy of eyewitness identification in a field setting. Journal of Personality and Social Psychology 42: 673.
- Wilson J, Hugenberg K, Bernstein M (2013) The Cross-Race Effect and Eyewitness Identification: How to Improve Recognition and Reduce Decision Errors in Eyewitness Situations. Social Issues and Policy Review 7: 83-113.
- Platz S, Hosch H (1988) Cross-Racial/Ethnic Eyewitness Identification: A Field Study. Journal of Applied Social Psychology 18: 972-984.
- Wixted JT, Wells GL(2017) The relationship between eyewitness confidence and identification accuracy: A new synthesis. Psychol Sci Public Interest 18: 10-65. [Crossref]
- Dawnay N, Stafford-Allen B, Moore D, Blackman S, Rendell P, et al. (2014) Developmental Validation of the ParaDNA® Screening System-A presumptive test for the detection of DNA on forensic evidence items. Forensic Sci Int Genet 11: 73-9. [Crossref]
- Blackman S, Dawnay N, Ball G, Stafford-Allen B, Tribble N, et al. (2015) Developmental validation of the ParaDNA® Intelligence System—A novel approach to DNA profiling. Forensic Sci Int Genet 17: 137-48. [Crossref]