Hereditary elliptocytosis (HE) is usually an autosomal dominant inherited hemolytic condition that is characterized by heterogeneous clinical course. Majority of the cases of HE are due to genetic defects affecting α-spectrin, β-spectrin or protein 4.1. Phenotypic expressivity can be different in different patient for single gene mutation i.e. different individuals with the same genotype can have more or less severe phenotype. I am reporting a heterozygous partial deletion in the EPB41 gene that was detected in a patient whose peripheral blood smear was consistent with HE and the deletion encompassed at least exons 10 to 16. This EPB41 deletion has not been previously reported in literature. Erythroid precursors in bone marrow of HE patients are round without any morphologic abnormalities which suggests elliptical shape of elliptocytes develop during the normal aging process of red blood cell (RBC) in circulation. The morphological changes seen in RBCs in HE do not necessarily shorten the lifespan of all patients with HE.
hereditary elliptocytosis, gene mutation, hemolysis, hereditary pyropoikilocytosis, anemia
Hereditary elliptocytosis (HE) is usually an autosomal dominant inherited hemolytic condition that is characterized by heterogeneous clinical course, ranging from an asymptomatic carrier to life-threatening condition requiring red cell transfusion [1]. Although the main mode of inheritance of HE is autosomal dominant however this condition can also be inherited as autosomal recessive condition as well as by novel mutations [2].
A 23 year old gentleman who was seen in haematology clinic for further management of an incidental finding of mild splenomegaly which was found on an ultrasound scan of abdomen that was performed to investigate intermittent jaundice. He informed me that he has been investigated for a while for recurrent episodes of jaundice and vomiting. He was found to have a fatty liver on an ultrasound scan of abdomen, gastritis on an upper gastrointestinal endoscopy and the ultrasound scan also shown enlarged spleen of 14.7 cm.
He generally feels tired and unwell. As he was adopted as a child he does not know about his parents. However, he reported he was told as a child that he had Gilbert's syndrome and he gets jaundiced every few months and usually just drinks lots of water until it settles. Apart from mental health issues he does not have other medical problem. He drinks alcohol less than once a week and very little amount. On examination there was no palpable lymphadenopathy.
His blood tests have confirmed a compensated haemolysis (Table 1) and microscopic examination of peripheral blood smear showed elliptocytes (Figure 1).
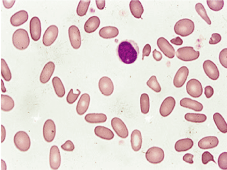
Figure 1. Peripheral blood film showing elliptocytes
His blood results are shown in (Table 1). Test for pathogenic variants of genes known to be associated with red cell membranopathy were performed using Agilent SureSelect enrichment technology and Illumina DNA sequencing. Reported variants were confirmed by dosage-sensitive polymerase chain reaction (PCR), Sanger sequencing and fragment analysis and results are shown in (Table 2).
Table 1. Blood result
Test |
Result |
Normal range |
Hb |
147 |
133-173 g/L |
MCV |
85.2 |
80-96 fl |
MCH |
31.1 |
27-32 pg |
MCHC |
360 |
320-360 g/L |
RDW |
16.5 |
9-15% |
Reticulocyte |
5.1 |
0.2-2% |
Total bilirubin |
45 |
1-21 umol/L |
Conjugated bilirubin |
15 |
0-5 umol/L |
LDH |
430 |
0-250 iu/L |
ALT |
40 |
0-41 iu/L |
Haptoglobin |
<0.3 |
0.5-2 g/L |
G6PD screen |
Normal result |
|
Hb: hemoglobin; MCV: mean corpuscular volume; MCH: mean corpuscular haemoglobin; MCHC: mean corpuscular hemoglobin concentration; RDW: red cell distribution width; LDH: lactate dehydrogenase; ALT: alanine transaminase; G6PD: glucose 6 phosphodehydrogenase
Table 2. Red cell gene panel result
Red Cell Gene Panel Result |
Heterozygous partial deletion in the EPB41 gene detected. Exact breakpoint is not known, however the deletion encompasses at least exons 10 to 16.
Interpretation: These variants are likely to be pathogenic and are consistent with a diagnosis of EPB41 associated HE. |
Following genes are included in analysis |
Membranopathy subpanel: ABCG5, ABCG8, ADD1, ADD2, AK1, ANK1, APOB, EPB41, EPB42, DMTN, KCNN4, MTTP, PIEZO1, RhAG, SLC2A1, SLC4A1, SPTA1, SPTB, STOM, TMOD1, TPM3, XK. |
Erythroid precursors in bone marrow of HE patients are round without any morphologic abnormalities which suggests elliptical shape of elliptocytes develop during the normal aging process of red blood cell (RBC) in circulation [3]. The mechanism involves repeated episodes of deformation that occur as RBCs goes through capillary beds; repeated deformation causes permanent damages and changes in the cytoskeleton of RBCs in HE. [4] Normal RBCs recovers their normal biconcave physical shape after they emerge from narrow capillaries. However, RBCs from patient with HE seems to lack normal interactions between cytoskeletal and membrane components that cause the cells gain an elliptocytic shape.
Majority of the cases of HE are due to genetic defects affecting α-spectrin, β-spectrin or protein 4.1 [5]. Variants in alpha-spectrin is the commonest genetic defect affecting 65% of HE patients and abnormalities in beta-spectrin are accounting for approximately 30 percent of cases. Protein 4.1 abnormalities affects approximately 5 percent of HE cases [6]. Phenotypic expressivity can be different in different patient for single gene mutation i.e. different individuals with the same genotype can have more or less severe phenotype. Heterozygotes are usually asymptomatic with mild or no hemolysis, whereas homozygotes or compound heterozygotes often have symptomatic hemolytic anemia.
The morphological changes seen in RBCs in HE do not necessarily shorten the lifespan of all patients with HE as it evidenced by the observation of a normal lifespan of RBCs from patients with Southeast Asian ovalocytosis (SAO; caused by band 3 protein abnormalities) despite the oval shape of these cells as well increased rigidity of RBC membrane [3,7,8] Hence patients with SAO usually do not have hemolytic anemia. Reduced membrane stability can lead to RBC fragmentation and this can lead to production of microcytic or spherocytic RBCs. The severity of hemolysis relates to the amount of membrane loss. [1,3] RBCs in hereditary pyropoikilocytosis (HPP)-the autosomal recessive variant of HE are susceptible to RBC budding and fragmentation upon heating to 46°C, whereas normal RBCs are not affected by temperatures below 50°C [9,10].
M S Islam conceived and design the manuscript, written the manuscript, managed the patient, collected data, performed the literature search.
Talhah Saad Bin-Islam for editing the manuscript and critical comments.
None.
None to disclose.
- An X, Mohandas N (2008) Disorders of red cell membrane. Br J Haematol 141: 367-375. [Crossref]
- Omar Nissa, Satheesh Chonat, Neha Dagaonkar, Marya O (2016) Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol Dis 61: 4-9. [Crossref]
- Palek J, Jarolim P (1993) Clinical expression and laboratory detection of red blood cell membrane protein mutations. Semin Hematol 30: 249-283. [Crossref]
- Tomaselli MB, John KM, Lux SE (1981) Elliptical erythrocyte membrane skeletons and heat-sensitive spectrin in hereditary elliptocytosis. Proc Natl Acad Sci USA 78: 1911.
- Soderquist C, Bagg A (2013) Hereditary elliptocytosis. Blood 121: 3066. [Crossref]
- Gallagher PG (2005) Red cell membrane disorders. Hematology Am Soc Hematol Educ Program 2005: 13-18. [Crossref]
- Delaunay J, Dhermy D (1993) Mutations involving the spectrin heterodimer contact site: clinical expression and alterations in specific function. Semin Hematol 30: 21-33. [Crossref]
- Lecomte MC, Garbarz M, Gautero H (1993) Molecular basis of clinical and morphological heterogeneity in hereditary elliptocytosis (HE) with spectrin alpha I variants. Br J Haematol 85: 584.
- Zarkowsky HS, Mohandas N, Speaker CB, Shohet SBA (1975) A congenital haemolytic anaemia with thermal sensitivity of the erythrocyte membrane. Br J Haematol 29: 537.
- Grace RF, Lux S (2009) Disorders of the red cell membrane. In: Orkin SH, Nathan DG, Ginsburg D (Eds), (7th Edn) Hematology of Infancy and Childhood, Saunders, Philadelphia. p. 659.