Otitis media (OM) is the most common bacterial infection in children, and often leads to conductive hearing loss. The Gram-negative bacterial pathogen, nontypeable Haemophilus influenzae (NTHi) is one of the primary causative agents. A classic hallmark of NTHi-induced OM is inflammation in middle ear. While appropriate inflammation is critical for host defense, an overactive response can often be detrimental to the host. Thus, inflammation must be tightly controlled. Here, we show that NTHi up-regulates the expression of granulocyte macrophage-colony stimulating factor (GM-CSF), one of the major proinflammatory mediator, via MEK-ERK signaling pathway in human middle ear epithelial cells. Moreover, cAMP-dependent protein kinase A (PKA) negatively regulates NTHi-induced GM-CSF expression through inhibition of MEK-ERK signaling pathway. Collectively, our studies unveil a new mechanism underlying the tight regulation of GM-CSF expression via a negative cross-talk between cAMP-PKA and MEK-ERK pathways and may shed light on developing new anti-inflammatory strategies.
Nontypeable Haemophilus influenzae, GM-CSF, negative regulation, ERK, PKA
Otitis media (OM) is the most common pediatric infectious disease. Since this disease often leads to conductive hearing loss during the crucial period of speech and language development, children with OM may suffer speech and language disabilities [1,2]. Nontypeable Haemophilus influenzae (NTHi) represents one of the most common Gram-negative bacterial pathogen causing OM [3]. Antibiotics are frequently used for the treatment of OM. However, inappropriate antibiotic treatment contributes significantly to the increased multidrug-resistant strains [4,5]. Thus, development of new therapeutic strategies is needed for the treatment of OM based on understanding of its molecular pathogenesis.
An excessive inflammation caused by bacterial infection is a hallmark of OM, characterized by up-regulated proinflammatory mediators [6,7]. Inflammation plays an important role in host defense against bacteria. However, if uncontrolled, excessive inflammation often causes tissue damage and immunopathology such as OM [8,9]. Thus, inflammation must be tightly regulated. Among various proinflammatory mediators, granulocyte macrophage colony-stimulating factor (GM-CSF) plays a critical role in stimulating proliferation and differentiation of granulocytes and macrophages from progenitor cells, and is thus a critical proinflammatory mediator for the host response to infection [10]. The suppression of lipopolysaccharide (LPS)-induced inflammation was observed in GM-CSF-deficient mice [11]. Consistent with this finding, GM-CSF administration in mice led to enhanced up-regulation of proinflammatory cytokines in response to LPS and tumor necrosis factor-α (TNF-α) [12]. Moreover, GM-CSF is identified in middle ear effusion from OM patients [13]. However, the molecular mechanisms underlying the tight regulation of GM-CSF induction by NTHi remain largely unknown. Understanding the regulatory mechanism of NTHi-induced GM-CSF expression in middle ear may lead to developing new therapeutic strategies for OM.
Cyclic adenosine monophosphate (cAMP) is a second messenger that has been shown to regulate inflammation and immune response [14-18]. Previous studies have shown that cAMP regulates multiple signal pathways via a major downstream effector protein kinase A (PKA) [15]. Increased intracellular cAMP levels in LPS-treated macrophages results in a suppression of proinflammatory cytokines such as TNF-α and IL-1β [19-21]. Thus, activating cAMP signaling may represent an interesting therapeutic strategy to suppress inflammation. Indeed, increasing intracellular cAMP by using phosphodiesterase 4B (PDE4B) inhibitor roflumilast has been recently approved for treating severe chronic obstructive pulmonary disease (COPD) [22]. The role of cAMP-dependent PKA signaling pathway in NTHi-induced GM-CSF expression in middle ear has yet to be investigated.
In the present study, we investigated the role and underlying molecular mechanism of cAMP- PKA pathway in NTHi-induced GM-CSF expression in human middle ear epithelial cells. We found that NTHi induces GM-CSF expression via MEK-ERK signaling pathway. We further showed that elevated cAMP suppresses NTHi-induced GM-CSF expression through inhibition of ERK activation. Moreover, we demonstrated that cAMP-dependent PKA is involved in negatively regulating NTHi-induced GM-CSF expression. Thus, our study provides the direct evidence for the first time for the negative regulation of GM-CSF induction by NTHi via cAMP-dependent PKA signaling pathway and may help develop new anti-inflammatory therapeutic strategies.
Reagents and antibodies
PD98059, U0126, forskolin, and H89 were purchased from Enzo Life Sciences. N6-Phenyl-cAMP (6-Phe-cAMP) was purchased from BioLog. Antibodies for phospho-ERK1/2 (#9101) and total ERK1/2 (#9102) were purchased from Cell Signaling.
Bacterial strains and culture condition
Clinical isolates of NTHi strain 12, 2627, and 9274 were used in this study. NTHi were grown on chocolate agar plate at 37°C in an atmosphere of 5% CO2 overnight, and inoculated in brain heart infusion (BHI) broth supplemented with 3.5 μg/ml NAD and 10 μg/ml hemoglobin (BD Biosciences). After overnight incubation, bacteria were subcultured into fresh BHI and the log phase NTHi, monitored by measurement of optical density value, was washed and suspended in phosphate-buffered saline (PBS) for in vitro cell experiments and for in vivo animal experiments.
Cell culture
All media described below were supplemented with 10% (vol/vol) FBS (Sigma-Aldrich). Human middle ear epithelial HMEEC-1 cells were maintained in DMEM (Cellgro) supplemented with BEGM SingleQuots (Lonza). Human lung epithelial A549 cells were maintained in F-12K medium (Gibco). Human cervical epithelial HeLa cells were maintained in MEM medium (Cellgro).
Real-time quantitative RT-PCR analysis
Total RNA was isolated with TRIzol reagent (Life Technologies) by following the manufacturer’s instruction. For the reverse transcription reaction, TaqMan reverse transcription reagents (Life Technologies) were used as described previously [23,24]. For quantitative RT-PCR analysis, PCR amplifications were performed by using SYBR Green Universal Master Mix (Life Technologies). In brief, the reactions were performed in triplicate containing 2× Universal Master Mix, 1 μl of template cDNA, 200 nM primers in a final volume of 12.5 μl and they were analyzed in a 96-well optical reaction plate (USA Scientific). Reactions were amplified and quantified by using a StepOnePlus Real-Time PCR System and the manufacturer’s corresponding software (StepOnePlus Software v2.3; Life Technologies). The relative quantities of mRNAs were determined by using the comparative Ct method and were normalized by using human cyclophilin for in vitro or mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for in vivo as an endogenous control. The primers for human GM-CSF and cyclophilin were described previously [25]. The primer sequences for mouse GM-CSF and GAPDH are as follows: mouse GM-CSF (Forward 5’-ATGCCTGTCACGTTGAATGAAG-3’ and Reverse 5’-GCGGGTCTGCACACATGTTA-3’) and mouse GAPDH (Forward 5’-ACCCAGAAGACTGTGGATGG-3’ and Reverse 5’-GGATGCAGGGATGATGTTCT-3’.)
Plasmids and transfections
The expression plasmid of a constitutively active form of MEK (MEK CA) was described previously [26]. All transient transfections were carried out using TransIT-2020 reagent (Mirus) according to the manufacturer’s instructions.
Enzyme-linked immunosorbent assay (ELISA)
Cells were stimulated with NTHi for 12 h. Cell culture supernatants were harvested and centrifuged at 15,000 × g for 10 min to remove debris prior to analysis. Human GM-CSF protein was measured with PeproTech Human GM-CSF ELISA Development kit (PeproTech) according to the manufacturer’s instructions.
Western blot
Western blots were performed using whole-cell extracts in the lysis buffer (20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 50 mM Na4P2O7, 30 mM NaF, 5 μM ZnCl2, 2 mM Iodoacetic acid, 1% Triton-X, supplemented with 1 mM Na3VO4 and PIC), separated on 10% (wt/vol) SDS-PAGE
gels, and transferred to polyvinylidene difluoride (PVDF) membrane (GE Healthcare Life Sciences). The membrane was blocked with 5% (wt/vol) non-fat dry milk in a solution of TBS containing 0.1% Tween 20 (TBS-T). The membrane was then incubated in a 1:1,000 dilution of a primary antibody in 5% (wt/vol) non-fat dry milk-TBS-T. After three washes in TBS-T, the membrane was incubated with 1:5,000 dilution of the corresponding secondary antibody in 5% (wt/vol) non-fat dry milk-TBS-T. Respective proteins were visualized by using Amersham ECL Prime Regent (GE Healthcare Life Sciences).
Mice and animal experiments
C57BL/6J mice (10-12 weeks old) were anesthetized and transtympanically inoculated with 1×107 colony-forming units (c.f.u.) of NTHi, and PBS was inoculated as control. The inoculated mice were then sacrificed 5 h after NTHi inoculation. Dissected mouse middle ears were subjected to total RNA extraction. All animal experiments were carried out in accordance with guidelines of, and were approved by, the Institutional Animal Care and Use Committee (IACUC) at Georgia State University.
Statistical analysis
All experiments were repeated at least three times. Data are shown as mean ± SD. Statistical analysis was analyzed by performing unpaired two-tailed Student’s t-test. P<0.05 was considered statistically significant.
NTHi induces GM-CSF expression in human epithelial cells in vitro and mouse middle ear in vivo
We have recently reported that NTHi, a major human pathogen of OM and exacerbation of COPD, induces up-regulation of GM-CSF expression in human bronchial epithelial BEAS-2B cells [25,27]. However, the molecular mechanisms underlying the negative regulation of induction of GM-CSF by NTHi remain largely unknown. We first examined whether NTHi induces GM-CSF expression in a number of human epithelial cell lines by performing quantitative PCR (Q-PCR) analysis. NTHi induced GM-CSF expression at mRNA level in human middle ear epithelial HMEEC-1 cells (Figure 1A), human lung epithelial A549 cells (Figure 1B), and human cervical epithelial HeLa cells (Figure 1C). Additionally, we determined that NTHi induced GM-CSF expression in a dose-dependent manner (Figure1D). To further determine the generalizability of our finding, we tested several NTHi strains for GM-CSF mRNA induction [28]. NTHi strain 2627 and 9274 also induced GM-CSF expression at mRNA level in HMEEC-1 cells (Figure1E). Furthermore, NTHi also induced the expression of GM-CSF at protein level in HMEEC-1 cells (Figure1F). Consistent with the in vitro data, NTHi also enhanced GM-CSF expression at mRNA level in the mouse middle ear (Figure 1G). Together, these data demonstrate that NTHi induces GM-CSF expression at both mRNA and protein levels in human epithelial cells in vitro and mouse middle ear in vivo.
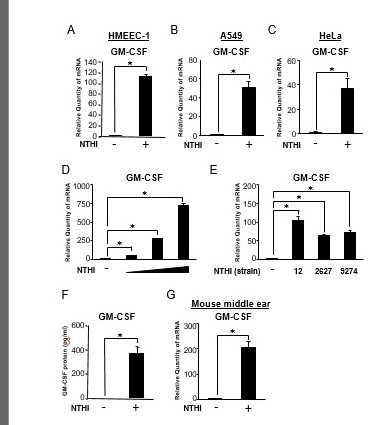
Figure 1.
NTHi induces GM-CSF expression in human epithelial cells in vitro and mouse middle ear in vivo. (A, B and C) HMEEC-1 (A), A549 (B), and HeLa (C) cells were stimulated with NTHi for 5h, and GM-CSF mRNA expression was analyzed by Q-PCR. (D) HMEEC-1 cells were stimulated with increasing doses of NTHi for 5h, and GM-CSF mRNA expression was analyzed by Q-PCR. (E) HMEEC-1 cells were stimulated with various strains of NTHi (12, 2627 and 9274) for 5h, and GM-CSF mRNA expression was analyzed by Q-PCR. (F) HMEEC-1 cells were stimulated with NTHi for 12h, and the protein level of GM-CSF in supernatants was analyzed by enzyme-linked immunosorbent assay. (G) C57BL/6J mice were transtympanically inoculated with NTHi (1×107 c.f.u. per ear) for 5h, and GM-CSF mRNA expression was analyzed by Q-PCR. Data are mean ± SD (n = 3); *P < 0.05.
MEK-ERK signaling is required for NTHi-induced GM-CSF expression in human middle ear epithelial cells
Because we previously demonstrated that the MAP kinase ERK is critical for the regulation of NTHi-induced inflammatory response [26,29], we first examined the effects of PD98059 and U0126, specific ERK inhibitors, on NTHi-induced GM-CSF expression by performing Q-PCR analysis. As shown in Figure 2A and 2B, ERK inhibitors significantly suppressed the enhancement of NTHi-induced GM-CSF expression at mRNA level in a dose-dependent manner, indicating that activation of ERK is required for NTHi-induced GM-CSF expression. We next sought to determine if activation of ERK is also sufficient GM-CSF induction by expressing a constitutively active form of MEK (MEK CA), which phosphorylates ERK and results in the activation of ERK. As expected, the expression of GM-CSF was markedly induced by expressing MEK CA in HMEEC-1 cells (Figure 2C). In addition, ERK inhibitor U0126 also markedly suppressed MEK CA-induced GM-CSF expression (Figure 2D). Taken together, these results suggest that MEK-ERK signaling is required for NTHi-induced GM-CSF expression in human middle ear epithelial cells.
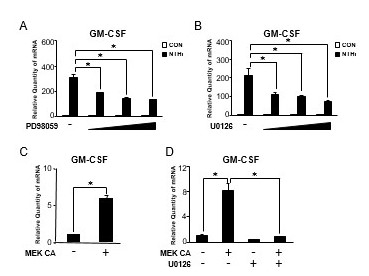
Figure 2.
MEK-ERK signaling pathway is required for NTHi-induced GM-CSF expression in human middle ear epithelial cells. (A) HMEEC-1 cells were pretreated with PD98059 (2.5, 5, and 10 μM) for 1 h followed by NTHi stimulation for 5 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (B) HMEEC-1 cells were pretreated with U0126 (1, 5, and 10 μM) for 1 h followed by NTHi stimulation for 5 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (C) HMEEC-1 cells were transfected with a constitutively active form of MEK (MEK CA) for 24 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (D) HMEEC-1 cells transfected with MEK CA for 24 h were treated with U0126 (10 μM) for 2 h, and GM-CSF mRNA expression was analyzed by Q-PCR. Data are mean ± SD (n = 3); *P < 0.05.
Elevated cAMP suppresses NTHi-induced GM-CSF expression via inhibition of ERK in human middle ear epithelial cells
cAMP, a key regulator of inflammatory and immune responses, has long been thought as a promising therapeutic target for inflammation [22,30]. Therefore, we first sought to determine the effect of cAMP in NTHi-induced GM-CSF expression by using forskolin, a potent activator of adenylate cyclase, which leads to an increase in the intracellular cAMP in human middle ear epithelial cells [31]. As shown in Figure 3A, forskolin significantly suppressed NTHi-induced GM-CSF expression in a dose-dependent manner in HMEEC-1 cells. Next, we sought to determine if cAMP elevated by forskolin regulates NTHi-induced ERK phosphorylation by performing western blot analysis. NTHi-induced ERK phosphorylation was markedly suppressed by forskolin in HMEEC-1 cells (Figure 3B). We further confirmed if forskolin can also suppress MEK CA-induced GM-CSF expression and ERK phosphorylation. As shown in Figure 3C and 3D, MEK CA-induced GM-CSF expression and ERK phosphorylation were significantly suppressed by forskolin. Together, these data demonstrate that elevated cAMP suppresses NTHi-induced GM-CSF expression via inhibition of ERK in human middle ear epithelial cells.
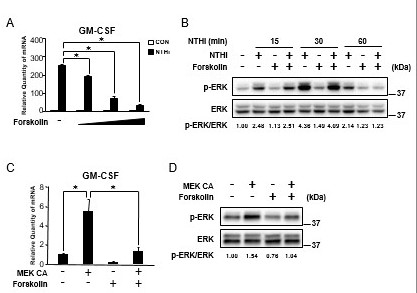
Figure 3.
Elevated cAMP suppresses NTHi-induced GM-CSF expression via inhibition of ERK in human middle ear epithelial cells. (A) HMEEC-1 cells were pretreated with forskolin (0.1, 1, and 10 μM) for 1 h followed by NTHi stimulation for 5 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (B) HMEEC-1 cells were pretreated with forskolin (1 μM) for 1 h followed by NTHi stimulation for indicated time, and cell lysates were analyzed by immunoblotting with the indicated antibodies. The fold change of phosphorylated ERK was quantified by densitometry and normalized to total ERK. (C) HMEEC-1 cells transfected with MEK CA for 24 h were treated with forskolin (1 μM) for 2 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (D) HMEEC-1 cells transfected with MEK CA for 24 h were treated with forskolin (1 μM) for 2 h, and cell lysates were analyzed by immunoblotting with the indicated antibodies. The fold change of phosphorylated ERK was quantified by densitometry and normalized to total ERK. Data in A and C are mean ± SD (n = 3); *P < 0.05.
2021 Copyright OAT. All rights reserv
PKA negatively regulates NTHi-induced GM-CSF expression via inhibition of MEK-ERK signaling pathway in human middle ear epithelial cells
Having shown that cAMP is involved in NTHi-induced GM-CSF expression in HMEEC-1 cells, we next investigated the role of the cAMP effector PKA [15]. As shown in Figure 4A, a selective PKA activator (6-Phe-cAMP) significantly suppressed NTHi-induced GM-CSF expression in a dose-dependent manner. This data indicates that cAMP-dependent activation of PKA is involved in the negative regulation of NTHi-induced GM-CSF expression. We further evaluated the involvement of PKA in NTHi-induced GM-CSF expression in HMEEC-1 cells treated with a selective PKA inhibitor (H89) and forskolin. As shown in Figure 4B, inhibition of NTHi-induced GM-CSF expression by forskolin was markedly reversed by H89. Next, we determined whether MEK CA-induced GM-CSF expression is regulated by PKA. As shown in Figure 4C, MEK CA-induced GM-CSF expression was significantly suppressed by PKA activator 6-Phe-cAMP. Taken together, PKA negatively regulates NTHi-induced GM-CSF expression via inhibition of MEK-ERK signaling pathway in human middle ear epithelial cells (Figure 4D).
In summary, our study demonstrates for the first time that cAMP-dependent PKA pathway acts as a negative regulator of MEK/ERK-mediated GM-CSF induction by NTHi. This finding is of particular translational interest as the MEK-ERK signaling pathway has long been thought as a promising therapeutic target. Understanding the intrinsic link between cAMP and ERK activation provides new therapeutic potentials to diseases in which ERK signaling is activated. It is important to note, however, that due to the critical role that the MEK-ERK signaling pathway plays on apoptosis and cell growth, extended manipulation of this pathway could lead to unwanted side effects. Therefore, treatments involving the manipulation of the MEK-ERK signaling pathway should be limited to short term usage. Future studies may provide a unique understanding of the nuances of NTHi-induced inflammation and provide direction in developing novel therapeutic strategies. Not only will this study provide critical understanding in controlling the immune response to a bacterial infection, but also its implications could be applied to other infectious and inflammatory diseases.
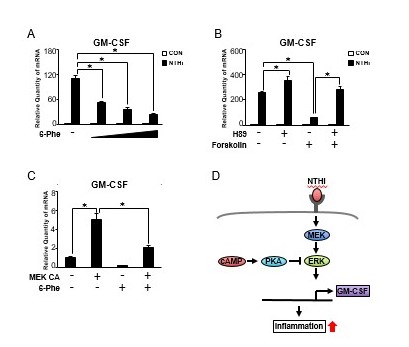
Figure 4.
PKA negatively regulates NTHi-induced GM-CSF expression via inhibition of MEK-ERK signaling pathway in human middle ear epithelial cells. (A) HMEEC-1 cells were pretreated with 6-Phe-cAMP (6-Phe, 0.1, 0.3, and 0.5 mM) for 1 h followed by NTHi stimulation for 5 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (B) HMEEC-1 cells were pretreated with H89 (20 μM) and forskolin (1 μM) for 1 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (C) HMEEC-1 cells transfected with MEK CA for 24 h were treated with 6-Phe-cAMP (0.5 mM) for 2 h, and GM-CSF mRNA expression was analyzed by Q-PCR. (D) A schematic diagram illustrating that cAMP-dependent PKA negatively regulates NTHi-induced GM-CSF expression via inhibition of MEK-ERK signaling pathway. Data are mean ± SD (n = 3); *P < 0.05.
This work was supported by National Institutes of Health Grants DC005843, DC004562, and DC013833 (to J.-D.L.), and Japan Society for the Promotion Science (JSPS) program on Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation (Grant Number S2510 to H.K.). J.-D.L. is Georgia Research Alliance Eminent Scholar in Inflammation and Immunity.
The authors declare no conflict of interest.
- Reichman J, Healey WC (1983) Learning disabilities and conductive hearing loss involving otitis media. J Learn Disabil 16: 272-278. [Crossref]
- Bluestone CD, Beery QC, Paradise JL (1973) Paradise, Audiometry and tympanometry in relation to middle ear effusions in children. Laryngoscope 83: 594-604.
- Faden H, Duffy L, Williams A, Krystofik DA, Wolf J (1995) Epidemiology of Nasopharyngeal Colonization with Nontypeable Haemophilus influenzae in the First 2 Years of Life. J Infect Dis 172: 132-135. [Crossref]
- Arguedas A, Kvaerner K, Liese J, Schilder AG, Pelton SI (2010) Otitis media across nine countries: disease burden and management. Int J Pediatr Otorhinolaryngol 74: 1419-1424. [Crossref]
- Ito M, Hotomi M, Maruyama Y, Hatano M, Sugimoto H, Yoshizaki T, et al, (2010) Clonal spread of ß-lactamase-producing amoxicillin–clavulanate-resistant (BLPACR) strains of non-typeable Haemophilus influenzae among young children attending a day care in Japan. Int J Pediatr Otorhinolaryngol 74: 901-906. [Crossref]
- MacArthur CJ, Hausman F, Kempton JB, Choi D, Trune DR (2013) Otitis media impacts hundreds of mouse middle and inner ear genes. PLoS One 8: e75213. [Crossref]
- Smirnova MG, Birchall JP, Pearson JP (2004) The immunoregulatory and allergy-associated cytokines in the aetiology of the otitis media with effusion. Mediators Inflamm 13: 75-88. [Crossref]
- Leichtle A, Lai Y, Wollenberg B, Wasserman SI, Ryan AF (2011) Innate signaling in otitis media: pathogenesis and recovery. Curr Allergy Asthma Rep 11: 78-84. [Crossref]
- Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454: 428-435. [Crossref]
- Hamilton JA (2008) Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8: 533-544. [Crossref]
- Basu S, Dunn AR, Marino MW, Savoia H, Hodgson G, Lieschke GJ, et al. (1997) Increased tolerance to endotoxin by granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol 159: 1412-1417. [Crossref]
- Brissette WH, Baker DA, Stam EJ, Umland JP, Griffiths RJ (1995) GM-CSF rapidly primes mice for enhanced cytokine production in response to LPS and TNF. Cytokine 7: 291-295. [Crossref]
- Himi T, Suzuki T, Kodama H, Takezawa H, Kataura A (1992) Immunologic characteristics of cytokines in otitis media with effusion. Ann Otol Rhinol Laryngol Suppl 157: 21-25.
- Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N (1996) Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem 271: 20828-20835.
- Cheng X, Ji Z, Tsalkova T, Mei F (2008) Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 40: 651-662. [Crossref]
- Hertz AL, Bender AT, Smith KC, Gilchrist M, Amieux PS, Aderem A, et al. (2009) Elevated cyclic AMP and PDE4 inhibition induce chemokine expression in human monocyte-derived macrophages, Proc Natl Acad Sci USA 106: 21978-21983.
- Gerlo S, Kooijman R, Beck IM, Kolmus K, Spooren A, et al. (2011) Cyclic AMP: a selective modulator of NF-IºB action. Cell Mol Life Sci 68: 3823-3841. [Crossref]
- Oldenburger A, Roscioni SS, Jansen E, Menzen MH, Halayko AJ, et al. (2012) Anti-inflammatory role of the cAMP effectors Epac and PKA: implications in chronic obstructive pulmonary disease. PLoS One 7: e31574. [Crossref]
- Seldon PM, Barnes PJ, Meja K, Giembycz MA (1995) Suppression of lipopolysaccharide-induced tumor necrosis factor-alpha generation from human peripheral blood monocytes by inhibitors of phosphodiesterase 4: interaction with stimulants of adenylyl cyclase. Mol Pharmacol 48: 747-757.
- Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D, et al. (1988) Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J Biol Chem 263: 5380-5384.
- Gerlo S, Verdood P, Kooijman R (2010) Modulation of cytokine production by cyclic adenosine monophosphate analogs in human leukocytes. J Interferon Cytokine Res 30: 883-891.
- Raker VK, Becker C, Steinbrink K (2016) The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front Immunol 7: 123. [Crossref]
- Komatsu K, Lee JY, Miyata M, Hyang Lim J, Jono H, et al. (2013) Inhibition of PDE4B suppresses inflammation by increasing expression of the deubiquitinase CYLD. Nat Commun 4: 1684. [Crossref]
- Miyata M, Lee JY, Susuki-Miyata S, Wang WY, Xu H1, et al. (2015) Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat Commun 6: 6062. [Crossref]
- Susuki-Miyata S, Miyata M, Lee BC, Xu H, Kai H, et al. (2015) Cross-talk between PKA-Cβ and p65 mediates synergistic induction of PDE4B by roflumilast and NTHi. Proc Natl Acad Sci U S A 112: E1800-1809. [Crossref]
- Wang WY, Komatsu K, Huang Y, Wu J, Zhang W, Lee JY, et al. (2014) CYLD negatively regulates nontypeable Haemophilus influenzae-induced IL-8 expression via phosphatase MKP-1-dependent inhibition of ERK. PLoS ONE 9: e112516- e1125169.
- Andrews CS, Matsuyama S, Lee BC, Li JD (2016) Resveratrol suppresses NTHi-induced inflammation via up-regulation of the negative regulator MyD88 short. Sci Rep 6: 34445. [Crossref]
- Gu XX, Tsai CM, Apicella MA, Lim DJ (1995) Quantitation and biological properties of released and cell-bound lipooligosaccharides from nontypeable Haemophilus influenza. Infect Immun 63: 4115-4120. [Crossref]
- Wang B, Cleary PP, Xu H, Li JD (2003) Up-regulation of interleukin-8 by novel small cytoplasmic molecules of nontypeable Haemophilus influenzae via p38 and extracellular signal-regulated kinase pathways. Infect Immun 71: 5523-5530. [Crossref]
- Kaur M, Holden NM, Wilson SM, Sukkar MB, Chung KF, Barnes PF, et al. (2008) Effect of 2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. AJP: Lung Cellular and Molecular Physiology 295: L505-L514.
- Laurenza A, Sutkowski EM, Seamon KB (1989) Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol Sci 10: 442-447.