An increase in the cholesterol content in phospholipid bilayer membranes is known to engender high order structures of lipids, which involve straightened (extended) lipid acyl chains. Lipid rafts are rich in such extended chains and considered to help clustering/multimerization of transmembrane helical peptides. Our recent atomistic simulations showed that the dimeric state of model helical peptides is stabilized in raft-like bilayers and that the potential energy term ascribed to lipid-peptide interactions contributes to the stabilization. Here our computation shows that the number of those lipids atoms which simultaneously contacted with both helical peptides in the dimeric state (which we refer to as 'dual contacts') was greater for a raft-like bilayer (1:1:1 palmitoyloleoylphosphatidylcholine (POPC)/dipalmitoylphosphatidylcholine (DPPC)/cholesterol bilayer) compared to a dioleoylphosphatidylcholine (DOPC) bilayer. Specifically, the distal half (i.e., near the CH3-terminal) of the acyl chains as well as the phospholipids head groups showed such differences. The number of the lipid atoms with such dual contacts was associated with the unsigned value of lipid-peptide term of the potential energy. Thus, the extended structures of saturated acyl chains in raft-like bilayers appear to enable phospholipids molecules to reside in the groove between, and contact with, both peptides in the dimeric/multimeric state, thereby modulating the potential energy in favor of the dimeric/multimeric state in a sequence-nonspecific manner. The formation of such dual contacts is likely to be assisted by small tilt and helix-helix crossing angles of the dimerized peptides which also appear to be brought about by extended acyl chains in the raft-like bilayer.
cholesterol, transmembrane helical peptides, lipids
It has been widely accepted that dimerization or multimerization of transmembrane (TM) domains of membrane proteins plays important roles in regulations of signal transduction. In particular, recent studies on single-pass receptor proteins have elucidated that dimerization of TM domains is a prerequisite step for receptor activation, although activation has been shown in many cases to require further structural changes after dimerization [1,2].
Among the factors tuning the propensity of membrane protein clustering, lipid environment has significant influences on the monomer-dimer equilibrium and can modulate the density of the dimerized receptor proteins [3]. However, compared to specific interactions such as those between cholesterol and cholesterol-recognizing peptide motifs [4], sequence-nonspecific effects of cholesterol and FAs on peptide dimerization have been addressed in a limited number of studies. Sequence-nonspecific effects could be important to set the basal activities of receptor proteins, thereby adjusting the tone of cellular activities such as intensity of inflammation [3]. Yano et al [5] showed that homodimer of a helical peptide with a sequence of (AALALAA)3 is stabilized in a 7:3 POPC/cholesterol bilayer compared to the POPC bilayer.
We recently performed a simulation analysis of self-dimerization using a model helical peptide ((Ile)21, which we refer to as the poly-Ile peptide) using the GROMOS united-atom parameter set and reported that the peptide tends to self-associates (dimerizes) in a 1:1:1 POPC/DPPC/cholesterol bilayer (which we henceforth refer to as the raft-like bilayer) but such propensity is negligible in a DOPC bilayer membrane [6]. Our free energy computation quantified the difference in the dimerization propensity between the two bilayers. With most current simulation parameters suitable for biomembranes, the potential energy is defined as the sum of the Lennard-Jones (LJ) energy VLJ and the electrostatic (Coulombic) energy VCoul, their combination accounting for the interactions of basically all pairs of atoms in the simulation system. We further reported that, when the total potential energy V was decomposed into the lipid-lipid, the peptide-peptide and the lipid-peptide interaction terms (which we represent as Vlipid-lipid, Vpept-pept, and Vlipid-pept, respectively), Vlipid-pept was found to be the key factor driving the stabilization of the dimeric state of the poly-Ile peptide in the raft-like bilayer [6]. Contrary to our early hypothesis that the tight cholesterol-phospholipid interactions might act to exclude and segregate peptides and therefore that the Vlipid-lipid potential energy term might be the key contributor, the Vlipid-lipid term did not show a clear contribution to the increased stability of the peptide dimer in the raft-like 1:1:1 bilayer [6]. Our further decomposition showed that, both VLJlipid-pept and VCoullipid-pept (i.e., the LJ and Coulombic (electrostatic) components of Vlipid-pept) serve as the factors driving the dimer stabilization in the raft-like bilayer.
Then, one may ask the following question. Why and how do VLJlipid-pept and VCoullipid-pept change depending on the inter-helical distance r in favor of the dimer stabilization in raft-like bilayers? In other words, what is the structural basis underlying the changes in these Vlipid-pept terms that occur upon, and contributes to, the dimerization in raft-like bilayers? More specifically, is there any advantage for the straightened (extended) structure of phospholipids in solvating the peptide dimer compared to the monomers [3]? In this study, we focused on VLJlipid-pept, i.e., the LJ component of the lipid-peptide interaction potential energy, in an attempt to find structural features of the extended lipids of the raft-like bilayers in contact with peptides in comparison with the lipids in the non-raft DOPC bilayer.
For simulations, Gromacs suite version 4.5.4 was used [7]. For the simulation parameter set, the GROMOS53A6 united-atom force field was used [8,9]. The DOPC, POPC, dipalmitoylphosphatidylcholine (DPPC) and cholesterol topology parameters implemented the Gromacs were used. For the water molecule, the simple-point charge (SPC) water [10] was used. The initial structures (.gro files) of the lipid bilayers were obtained from our recent files [11]. The initial structures of the poly-Ile peptides ((Ile)21) in the antiparallel configuration were sampled from our recent study [6] and the N- and C-termini were capped with a acetyl and NH2 groups, respectively.
Run parameters were set as follows. Semi-isotropic coupling was used to set the pressure at 1 bar. For treatment of the long-range electrostatic energy, PME was used. For contact analysis, the eight simulation trajectories (100ns equilibration runs followed by 400ns productive runs) obtained from our study were used [6], but eight additional 300ns umbrella-sampling runs were performed with the inter-helical separation r restrained at 2.0 nm, given that the umbrella sampling covered only 1.1 to 1.6 nm in the latter study. Run parameters were basically set as we described [6]. To control the temperature at 323 K, the Berendsen thermostat was used [12]. The bond lengths of lipids and proteins were restrained with LINCS [13] and those of the water with SETTLE [14]. The particle-mesh Ewald algorithm [15] was used with a real-space cutoff of 1.4 nm and the minimal grid size of 0.12 nm.
The peptide-lipid contacts were analyzed using our self-made programs which were prepared with attention to the periodic boundary condition. Here we refer to an atom which was located within Dcutoff from both of the peptides as an atom with 'dual contacts'. Two different Dcutoff values (5 and 6Å) were used. We let Nlip_atomdual represent the number of the lipid atoms (including cholesterol also) that have dual contacts (i.e., those lipid atoms located within Dcutoff from both of peptides). We further use Nlip_2-6-atomdual and Nlip_7-11-atomdual, and Nlip_12-16-atomdual to represent the number of the dually-contacting atoms among those atoms comprising the lipid acyl chain segments C2-C6, C7-C11 and C12-C16, respectively. Likewise, Nlip_choloh-atomdual and Nlip_cholc-atomdual represent the dually-contacting atoms of the OH group and the remaining united-atom CH/CH2/CH3 cholesterol particles, respectively. Nlip_head-atomdual represents the dually-contacting atoms of phospholipids head groups, where head groups are defined to be comprised of the phosphorylcholine group and glycerol backbone including oxygen (ester and carbonyl) atoms as well as the carbon atom, C1, of the carboxyl group.
For the contact analysis of lipids acyl chains, we let the number Nacyldual represent the number of acyl chains whose all of CH2/CH3 particles had dual contacts with the peptides. As such acyl chains are too few, we further introduced Nacyl-proxdual to denote the number of the acyl chains whose all of C2-C9 atoms had dual contacts. Similarly, we let Nacyl-distdual represent the number of acyl chains whose C9-C16 particles all had dual contacts.
For the orientation analysis, the 'C-P orientation' was defined as the angle between the z-axis (i.e., the direction of the bilayer normal) and the vector linking the middle carbon atom of the glycerol backbone to the phosphorus atom of the phosphatidylcholine. Similarly, the 'C-N orientation' was defined using the vector ending at the nitrogen atom of the choline group. The peptide tilt angle was computed using the program g_helixorient of Gromacs that uses the coordinates of four consecutive C atoms to define the local direction of helix axis. We represented the helix tilt angle against the z-axis based on the average of the four residue-segments of the C7-C14 segment.
Our recent simulations showed that the lipid raft-like bilayer (1:1:1 POPC/DPPC/cholesterol bilayer) stabilizes the dimeric state of the poly-Ile model peptide (Ile)21 compared to the DOPC bilayer [6]. In the former bilayer, POPC and DPPC molecules were the lipid species that were directly contacting with the peptides, whereas the atoms of cholesterol were seldom located within 3Å from the peptide surface. When we decomposed the potential energy into the peptide-peptide, lipid-lipid and lipid-peptide terms (Vpept-pept, Vlipid-lipid, and Vlipid-pept, respectively), the lipid-peptide term Vlipid-pept (i.e., the sum of the potential energies from all peptide atom-lipid atom pairs) was found to be an important component based on the finding that it changed as a function of the inter-helical distance r in favor of the peptide dimer stabilization in the raft-like 1:1:1 bilayer [6]. We then hypothesized that the ordered (extended) structure of acyl chains of the raft-like 1:1:1 bilayer may have some advantage in solvation of the dimerized peptide compared to the less-ordered acyl chains of the DOPC bilayer [3].
Dual contacts of acyl chains to dimeric peptides are relatively frequent in the raft-like bilayer compared to the DOPC bilayer
To gain insights into atomistic details of acyl chains-peptides contacts, we counted the number of phospholipids acyl chains that were in contact with peptides. We refer to a lipid atom as a 'dually-contacting' atom when it has contacts with both peptides simultaneously. We first counted the number, Nacyldual, of those acyl chains whose CH2 and CH3 particles (united-atoms, in this study) all had dual contacts, but such chains were too few (data not shown). When we counted the number, Nacyl-proxdual, of those acyl chains in which all of the eight proximal (i.e., C2-C9) CH2 particles had dual contacts to the dimeric peptides (under the criteria of <5Å from the nearest peptide atom), the count was similar between the DOPC (0.097) and the raft-like 1:1:1 bilayer (0.088) (Table 1). Nonetheless, when the dual contacts of all particles of the distal segment (C9-C16) were used as the criteria instead, the count (Nacyl-distdual) was greater for the raft-like 1:1:1 bilayer (0.170) than for the non-raft-type DOPC bilayer (0.089) (p = 0.001 based on eight independent trajectories) (Table 1). Some examples of the distal acyl chain segments that had dually-contacting atoms are demonstrated in Figure 1. When the definition of contact was loosened to '<6Å from the nearest peptide atom', the difference between the raft-like and non-raft bilayers was less pronounced, but there was a similar difference in Nacyl-distdual between the two bilayers (p = 0.004) (Table 1). When the peptides were in the monomeric state (r = 2.0 nm), neither the proximal nor distal segments of acyl chains showed an appreciable level of dual contacts as expected (Table 1). These results suggested that extended acyl chains and, more specifically, their distal portions (atoms closer to the CH3 end) in the raft-like bilayers are more likely to have dual contacts with both of the dimerized peptides compared to the corresponding atoms of the chains in the non-raft type bilayers.
Table 1 Summary of lipid-peptide potential energy and the number of acyl chains in dual contacts with peptides. This table shows mean ± SD, but for VLJlipid-pept mean ± SE is shown. *p values on comparison with the corresponding value in the DOPC bilayer are shown. All comparisons between the bilayers were statistically insignificant at r = 2.0 nm.
bilayer |
DOPC |
1:1:1 raft-like |
r (nm) |
1.3 (dimer) |
2.0 (monomer) |
1.3 (dimer)* |
2.0 (monomer) |
VLJlipid-pept |
-1281.4 ± 13.2 |
-1420.9 ± 4.5 |
-1272.7 ± 7.4 |
-1396.7 ± 5.6 |
VLJlipid-pept mean relative to r = 2.0 |
139.5 |
0 |
124.0 |
0 |
Nacyl-proxdual, (Dcutoff = 5Å) |
0.097 ± 0.309 |
0.011 ± 0.104 |
0.088 ± 0.293 (p = 0.573) |
0.008 ± 0.091 |
Nacyl-distdual, (Dcutoff = 5Å) |
0.089 ± 0.292 |
0.004 ± 0.067 |
0.170 ± 0.408 (p = 0.001) |
0.002 ± 0.046 |
Nacyl-proxdual, (Dcutoff = 6Å) |
1.106 ± 0.919 |
0.282 ± 0.513 |
1.215 ± 0.939 (p = 0.358) |
0.194 ± 0.434 |
Nacyl-distdual, (Dcutoff = 6Å) |
1.233 ± 0.934 |
0.115 ± 0.357 |
1.549 ± 0.991 (p = 0.004) |
0.090 ± 0.295 |
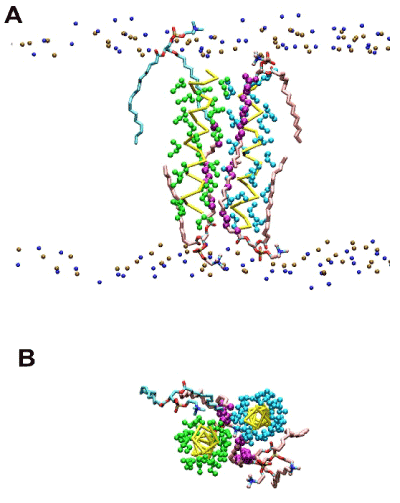
Figure 1. Representative snapshot exhibiting dually-contacting acyl chains in the raft-like 1:1:1 bilayer simulations of the poly-Ile peptides restrained at r = 1.3 nm (dimeric state). (A) Side view. Representation scheme: small spheres in top and bottom (ochre and blue), phospholipid head group atoms (phosphorus and nitrogen atoms, respectively); yellow bars (traces), peptide backbones; green and cyan spheres, Ile side chains; pink licorice, acyl chains of DPPC harboring dually-contacting atoms; cyan licorice, acyl chains of POPC with dually-contacting acyl chain(s); purple spheres, acyl chain CH2 (united) atoms that had dual contacts. (B) The same snapshot as (A) but a view from the top is shown. Lipids phosphorus and nitrogen atoms are hidden.
As the number of dually-contacting atom increases, the unsigned value of peptide-lipid LJ potential energy term increases, in favor of peptides dimerization
VLJlipid-pept (r) profile understandably shows a VLJlipid-pept (1.3) >VLJlipid-pept (2.0) difference due to the energy cost for desolvation; the dimerization (transition from r = 2.0 to 1.3 nm) requires detachment of several lipid molecules from the peptides (desolvation) causing the energy cost in the lipid-peptide term of the potential energy. As shown in Table 1 ('VLJlipid-pept relative to r = 2.0'), in the 1:1:1 bilayer, the LJ potential energy between the lipids and the peptides at r = 1.3 nm, i.e., VLJlipid-pept (1.3), relative to VLJlipid-pept (2.0) was not so high compared to the corresponding differential for the DOPC bilayer simulations [6]. This lower cost for desolvation has been considered to be an important factor for the dimer stabilization in the raft-like bilayer [3]. The smaller increment of VLJlipid-pept upon the peptide dimerization in the 1:1:1 bilayer may be attributed to, or at least associated with, the high number of dual contacts atoms when the peptides are dimerized.
This consideration prompted us to ask how such lipids contacts with peptides can associate with the LJ potential energy. In theory, the lipid atoms with dual contacts should make greater contributions to the energy, efficiently lowering it to a more negative value. We first counted the number Nlip_atomdual of the lipid atoms with dual contacts, and compared it between the dimeric (r = 1.3 nm) and monomeric state (r = 2.0 nm) (Table 2). This metric showed somewhat different values between the DOPC (31.3) and the raft-like 1:1:1 bilayer (32.5), but with an insignificant difference (p = 0.284) likely due to the large between-trajectory variances, which normally occur in similar atomistic simulations (see below). When we divided this metric Nlip-atomdual into those for head groups, three segments of acyl chains (C2-C6, C7-C11 and C12-C16), and cholesterol molecules, the contact count for each showed the results presented in Table 2; as expected, at r = 1.3 nm (dimeric state), there was a trend of difference in the number of dually-contacting atoms with the order of the 1:1:1 bilayer >the DOPC bilayer for the C7-C11 and C12-C16 segments, although p-value was 0.128 and 0.240, respectively. For the proximal segment C2-C6, an opposite trend (DOPC >1:1:1) was seen, but its interpretation is not straightforward because the contribution by cholesterol (counted separately in Table 2) should obscure this comparison. Together with the results on the acyl chains (Table 1), these support the view that the distal segments tend form more dual contacts with the dimeric peptides in raft-like bilayers compared to non-raft type bilayers.
Another feature in Table 2 was that an unexpectedly large difference in the number Nlip_head-atomdual of the dually-contacting head group atoms between the DOPC and 1:1:1 bilayers at r = 1.3 nm with the count being 3.7 and 5.0, respectively (p = 0.077). All the contact data in Table 2 were obtained with Dcutoff = 5Å. When the definition of contact was loosened to Dcutoff = 6Å, the number Nlip_head-atomdual of dually-contacting head group atoms was 8.6 (DOPC) and 12.0 (1:1:1) (p = 0.036) at r = 1.3 nm (details not shown). Thus, the head group atoms of the 1:1:1 bilayer also have higher tendency to have dual contact with the dimeric peptides compared to the DOPC bilayer. Although statistically insignificant, the total number, Nlip_atomdual, of dually-contacting atoms of lipids was higher for the 1:1:1 bilayer compared to the DOPC bilayer (Table 2). On the other hand, the number, Nlip_atomsingle, of lipids atoms with single contact is similar between the two bilayers (both being ~410), and, when the differential between this and the value for the monomeric state (r = 2.0 nm) was taken, the DOPC bilayer showed a greater number of lipid atoms with single contact than the 1:1:1 bilayer (the bottom row of Table 2). Overall, these results showed that, for the 1:1:1 bilayer, the dimerized peptides tend to have more dually-contacting lipid atoms compared to the monomeric state whereas the DOPC bilayer show a relatively modest level of this difference upon the peptide dimerization. The number of the lipid atoms with single contact (i.e., contacts with any atom(s) of only one peptide molecule) showed a marked decrease upon the peptide dimerization especially for the 1:1:1 bilayer. These findings were also consistent with the view that such a dimerization-associated increase of dual contacts associates with a reduction of the LJ potential energy.
Table 2. Analysis of atoms with dual contacts (mean ± SD). 1) For the oleoyl chains of DOPC and POPC, Nlip_17-18-atomdual, that is, the number of the dually-contacting atoms of the terminal two carbon atoms were also counted, but the results were, after adjustment, essentially similar to and inferable from the Nlip_12-16-atomdual values (details not shown). *Given the relatively high contact counts of cholesterol molecules, we considered statistical tests for these metrics to be irrelevant.
bilayer |
DOPC |
1:1:1 raft-like |
r (nm) |
1.3 (dimer) |
2.0 (monomer) |
1.3 (dimer) |
2.0 (monomer) |
VLJlipid-pept relative to r = 2.0 |
139.5 |
0 |
124.0 |
0 |
symbol |
analyzed group |
|
Nlip_head-atomdual |
PC head group |
3.7 ± 3.0 |
0.7 ± 1.6 |
5.0 ± 3.2 (p = 0.077) |
0.2 ± 0.8 |
Nlip_2-6-atomdual |
C2-C6 |
7.9 ± 3.4 |
2.4 ± 2.4 |
7.1 ± 3.3 (p = 0.111) |
1.3 ± 2.0 |
Nlip_7-11-atomdual |
C7-C11 |
7.9 ± 3.4 |
1.6 ± 2.1 |
8.4 ± 3.4 (p = 0.128) |
1.2 ± 1.7 |
Nlip_12-16-atomdual 1) |
C12-C16 |
7.9 ± 3.2 |
0.8 ± 1.5 |
8.7 ± 3.3 (p = 0.240) |
0.6 ± 1.2 |
Nlip_choloh-atomdual |
chol OH |
- |
- |
0.2 ± 0.6 |
0.1 ± 0.3 |
Nlip_cholc-atomdual |
chol carbon |
- |
- |
2.2 ± 3.4 |
0.5 ± 1.4 |
Nlip_atomdual |
total (all lipids) |
31.3 ± 6.8 |
5.6 ± 5.5 |
32.6 ± 6.9 (p = 0.284) |
4.0 ± 4.8 |
Nlip_head-atomsingle |
PC head group |
109.3 ± 15.5 |
143.5 ± 17.7 |
95.1 ± 14.0* |
111.4 ± 15.3 |
Nlip_2-6-atomsingle |
C2-C6 |
88.4 ± 9.3 |
112.0 ± 10.9 |
77.8 ± 9.0* |
94.9 ± 10.8 |
Nlip_7-11-atomsingle |
C7-C11 |
89.3 ± 9.7 |
107.9 ± 10.9 |
81.2 ± 10.2* |
103.9 ± 12.8 |
Nlip_12-16-atomsingle |
C12-C16 |
86.5 ± 9.7 |
100.9 ± 10.0 |
78.2 ± 11.1* |
103.4 ± 12.3 |
Nlip_choloh-atomsingle |
chol OH |
- |
- |
4.3 ± 2.7 |
6.4 ± 3.3 |
Nlip_cholc-atomsingle |
chol carbon |
- |
- |
65.6 ± 21.9 |
92.2 ± 27.6 |
Nlip_atomsingle |
total (all lipids) |
410.3 ± 25.0 |
509.1 ± 24.5 |
410.1 ± 26.0 |
523.0 ± 25.8 |
In the above, the eight 400 ns-trajectories at r =1.3 nm started from the independently prepared initial structures were used in order to avoid artifactual significances. Then, for these eight trajectories, we examined how the LJ potential energy associates with the number of the dually-contacting atoms. Strikingly, the number, Nlip-atomdual, showed a significant negative correlation with the potential energy term VLJlipid-pept with Pearson's correlation coefficient r = -0.813 and p = 0.014 (Table 3, Figure 2). This strongly suggests that the dual contacts of lipid atoms to the dimeric peptides are the main causative factor that brings down the VLJlipid-pept, thereby stabilizing the dimeric state of the peptides.
Table 3. The mean values of VLJlipid-pept and the contact count for the eight independent simulation trajectories of the 1:1:1 runs with r = 1.3 nm.
sim ID |
VLJlipid-pept |
Nlip-atomdual |
a |
-1299.1 |
36.15 |
b |
-1230.3 |
29.55 |
c |
-1277.9 |
32.33 |
d |
-1275.0 |
30.69 |
e |
-1285.3 |
33.22 |
f |
-1263.1 |
33.48 |
g |
-1278.7 |
34.52 |
h |
-12519 |
30.11 |
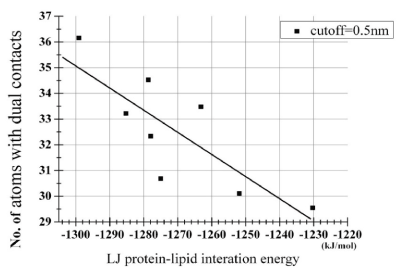
Figure 2. Scatterplot analysis of relationship between the peptide-lipid term of the LJ potential energy and the number of dually-contacting atoms shown in Table 3. The derived regression line was y = -0.087 x - 78.17. The correlation coefficient r between VLJlipid-pept and Nlip-atomdual was -0.813 (p = 0.014).
Lipid head groups have more dual contacts likely due to smaller tilt and crossing angle of helical peptides in the raft-like bilayer relative to the DOPC bilayer
Dimerization of TM helices is considered to be under the influences of several factors related to physicochemical properties of membranes and membrane-peptide hydrophobic mismatch. In the following we compare the basic structural properties of lipids and peptides between the two bilayers. Figure 3 compares the lipid order parameter -SCD between the two bilayers. In agreement with Niemela et al. [17], our 1:1:1 bilayer exhibited large unsigned values of -SCD, reminiscent of the high-order structure of the lipid rafts, although we have not analyzed sphingomyelin-containing membranes. To gain insights into the structure in the head group, we analyzed the C-P orientation, that is, the direction of the vector linking from the middle carbon atom of the glycerol backbone of phosphatidylcholine molecules to the phosphorus atom. Unexpectedly, the angle between the vector with the z-axis in the 1:1:1 raft-like bilayer was similar to that in the DOPC bilayer (Table 4). A similar trend was observed when the C-N orientation was analyzed (Table 4). These findings suggested that the overall structures of lipid head groups were similar between the bilayers. Therefore, the greater number of the dually-contacting head group atoms in the 1:1:1 bilayer shown in Table 2 should be ascribed not to structural features of head groups themselves but to some features of peptides including conformation and orientation.
Table 4. Basic structural properties of the used bilayer/peptides systems. 3)The length based on the distance between the Ile1 Cα-Ile21 Cα.
bilayer |
DOPC |
1:1:1 (raft-like) |
inter-helical distance r (nm) |
1.3 |
2.0 |
1.3 |
2.0 |
bilayer thickness based on P-P distance (nm) |
4.1 ± 0.1 |
4.0 ± 0.1 |
4.3 ± 0.1 |
4.3 ± 0.1 |
C-P tilt (degree) |
51.2 ± 3.5 |
48.0 ± 2.8 |
50.4 ± 2.7 |
50.8 ± 2.7 |
C-N tilt (degree) |
53.7 ± 2.5 |
52.2 ± 1.8 |
53.3 ± 2.1 |
52.9 ± 2.1 |
peptide tilt (degree) |
17.6 ± 9.6 |
17.5 ± 9.0 |
11.2 ± 5.2 |
11.5 ± 5.6 |
helix-helix
crossing angle (degree) |
26.7 ± 14.0 |
20.8 ± 11.1 |
14.8 ± 7.9 |
19.6 ± 17.0 |
peptide length3) (nm) |
3.30 ± 0.11 |
3.36 ± 0.14 |
3.38 ± 0.09 |
3.4 ± 0.1 |
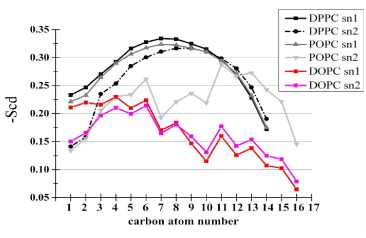
Figure 3. Lipid chain ordering within the bilayers. The ordering was quantified via the deuterium order parameter, -SCD, which is defined as -SCD = (1/2) Sz , where Sz = (1/2) «3cos2(qz) - 1», and qz stands for the angle between the C-H bond vector and the bilayer normal, and the double angle bracket denotes the ensemble average [16]. Sz can vary between 1 (full order along the normal) and -0.5 (full order perpendicular to the normal). The -SCD profiles of individual acyl chains of both bilayers are shown in one panel.
Table 1 Summary of lipid-peptide potential energy and the number of acyl chains in dual contacts with peptides. This table shows mean ± SD, but for VLJlipid-pept mean ± SE is shown. *p values on comparison with the corresponding value in the DOPC bilayer are shown. All comparisons between the bilayers were statistically insignificant at r = 2.0 nm.
The tilt angle of the peptides showed a pronounced difference between the bilayer, with that in the DOPC being greater than that in the 1:1:1 bilayer (Table 4). The helix-helix crossing angle was also small in the 1:1:1 relative to the DOPC bilayer, when the peptides were held at r = 1.3 nm (dimeric state). It is plausible that this relatively upright orientations of the peptides and small crossing angle facilitate the dual contacts of the lipid molecules in the 1:1:1 bilayer. Of note, it was not likely that these tilts and crossing angle differences were caused by the difference in the membrane thickness itself for the following reasons. First, the thickness was very similar between the DOPC and the 1:1:1 bilayers (Table 4). Moreover, unlike the hydrophobic peptides flanked by polar residues, both termini of the poly-Ile peptide used lack polar groups to anchor them to the lipid head groups. The latter feature led to an unappreciable degree of membrane deformation (for example, see Figure 1) and to the similar peptide lengths (Ile1 C-Ile21 C distance) between the DOPC and the 1:1:1 runs (Table 4), in support of a negligibly small effect of the negative mismatch on the dimerization. Further, the finding that the peptide-peptide potential energy profiles VCoulpept-pept and VLJpept-pept did not show features in favor of the dimer stabilization in the raft-like bilayer supports the idea that the tilt angle and the crossing angle are influential factors for the stabilization of the dimeric state through increasing the dual contacts but not through directly stabilizing the peptide-peptide associations [6]. Overall, in our setting, the tilt and crossing angles of peptides are likely to be an important factor for stabilization of the peptide dimer, but this effect appears to be mediated mainly by increasing the number of dually-contacting lipid atoms (of both acyl chains and head groups), not by impacting the peptide-membrane hydrophobic mismatch or by directly modulating the peptide-peptide interaction energy.
Our recent simulation analyses showed that the dimeric state of a model TM helical peptide (poly-Ile) was stabilized in the raft-like bilayer (1:1:1 POPC/DPPC/cholesterol bilayer) relative to the DOPC bilayer, and that the potential energy term Vlipid-pept that is ascribed to the lipid-peptide interactions plays a key role in the dimer stabilization [6]. To gain structural insights into the mechanisms by which the raft-like bilayer assists the peptide dimerization, we here analyzed the contacts between lipid and peptides in our simulation trajectories. When the two peptides were maintained in the dimerized state (with the inter-helical distance r = 1.3 nm), the number of those lipid atoms contacting with both peptides simultaneously (dual contacts) was greater in the raft-like bilayer (1:1:1 POPC/DPPC/cholesterol bilayer) compared to in the DOPC bilayer. Both the head groups and the distal, but not proximal, parts of acyl chains in the raft-like bilayers exhibited high propensity for such dual contacts to the dimerized peptides (Table 1).
Individual 400ns-simulation trajectories with the peptides held in the dimeric state showed a strong correlation between the number of the dually-contacting lipid atoms and the unsigned value of lipid-peptide term VLJlipid-pept of the LJ potential energy (Figure 2). Together with our recent findings, these suggest that the number of dual contacts is an important determinant for the potential energy term ascribed to the peptide-lipid interactions. Our findings also suggest that the straightened (extended) structure of acyl chains (those of saturated FAs or the chains straightened by cholesterol-phospholipid interaction) as well as the less tilted peptides orientation and the smaller helix-helix crossing angles in lipid-raft-type bilayers (Table 4) can jointly increase the dual contacts, thereby stabilizing the dimeric state of the peptides in a sequence-nonspecific manner. It is likely that extended and ordered acyl chains can increase the dual contacts through their own structures as well as through orienting the helical peptides toward the direction of the membrane normal, thereby increasing the chances for dual contacts.
However, the present study focused only on one peptide species and on the lipid-peptide LJ energy component. We have not focused on the electrostatic term VCoullipid-pept, given that, while its contribution to the dimer stabilization in the raft-like bilayer appeared plausible, the slow convergence of this value seemed to require longer simulations [6]. Further analyses with various peptides and bilayers are also necessary to better understand to what extent such dual contacts impact on the lipid-peptide potential energy, the total potential energy and the free energy for dimerization in more physiological settings.
The authors declare no competing financial interests.
2021 Copyright OAT. All rights reserv
- Sarabipour S (2017) Parallels and Distinctions in FGFR, VEGFR, and EGFR Mechanisms of Transmembrane Signaling. Biochemistry 56: 3159-3173. [Crossref]
- Bocharov EV, Mineev KS, Pavlov KV, Akimov SA, Kuznetsov AS, et al. (2017) Helix-helix interactions in membrane domains of bitopic proteins: Specificity and role of lipid environment. Biochim Biophys Acta 1859: 561-576. [Crossref]
- Nishizawa M, Nishizawa K (2017) Why have cholesterol and saturated fatty acids acquired proinflammatory roles during evolution? – A hypothesis from atomistic simulations showing sequence-nonspecific stabilization of peptide dimers in lipid raft-like bilayers Biomed Res Clin Prac 2: 1-6.
- Lange Y, Steck TL (2016) Active membrane cholesterol as a physiological effector. Chem Phys Lipids 199: 74-93. [Crossref]
- Yano Y, Kondo K, Kitani R, Yamamoto A, Matsuzaki K (2015) Cholesterol-induced lipophobic interaction between transmembrane helices using ensemble and single-molecule fluorescence resonance energy transfer. Biochemistry 54: 1371-1379. [Crossref]
- Nishizawa M, Nishizawa K (2017) Cholesterol and saturated fatty acid stabilize dimerization of helical transmembrane peptides by lowering energy cost related to peptides desolvation from lipids upon dimerization: an insight from atomistic simulation. Biomed Res Clin Prac 2: 1-8.
- Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 4: 435-447. [Crossref]
- Oostenbrink C, Villa A, Mark AE, van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25: 1656-1676. [Crossref]
- Poger D, Van Gunsteren WF, Mark AE (2010) A new force field for simulating phosphatidylcholine bilayers. J Comput Chem 6: 1117-1125. [Crossref]
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J (1981) Intermolecular Forces Interaction Models for Water in Relation to Protein Hydration (D. Reidel Publishing Co., Dordrecht, The Netherland, 1981) pp 331-342.
- Nishizawa M, Nishizawa K (2016) Free energy of helical transmembrane peptide dimerization in OPLS-AA/Berger force field simulations: inaccuracy and implications for partner-specific Lennard-Jones parameters between peptides and lipids. Molecular Simulation 42: 916-926.
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81: 3684.
- Hess B, Bekker H, Berendsen HJC (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18: 1463-1472.
- Miyamoto S, Kollman PA (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13: 952-962.
- Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys 98: 10089.
- Róg T, Pasenkiewicz-Gierula M, Vattulainen I, Karttunen M (2009) Ordering effects of cholesterol and its analogues. Biochim Biophys Acta 1788: 97-121. [Crossref]
- Niemelä PS, Hyvönen MT, Vattulainen I (2009) Atom-scale molecular interactions in lipid raft mixtures. Biochim Biophys Acta 1788: 122-135. [Crossref]