Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide. While other diseases associated with smoking have entered a plateau with respect to mortality, mortality associated with COPD has been increasing. Several facts seem to explain this phenomenon such as the increase in smoking habits in developing countries, the increase in life expectancy in high-income countries, and the growing increase in COPD not associated with smoking. A very striking finding of the entity is that the deterioration in lung function and therefore in mortality, is not stopped by the fact of suspending exposure to the environmental noxa. This could also contribute to this increase in mortality. The explanations considered are two: frequent respiratory infections in these patients, which are the leading cause of exacerbations, generate endogenous oxidative stress due to mitochondrial respiration, which perpetuates the inflammatory process, although exogenous exposure is suspended. The other explanation is autoimmunity playing a pathogenic role in the development and evolution of COPD. This monograph, therefore, aims to review the evidence of the immunological phenomenon in COPD giving special emphasis to two aspects: autoimmunity generating COPD and the possible therapeutic avenues that can be derived from this perspective. This last aspect seems crucial, especially since the current treatment does not prevent the deterioration of lung function or reduce mortality. At the end, some suggestions are made about future research.
chronic obstructive pulmonary disease (COPD), autoimmunity, autoantibodies, self-regulating T cells
COPD is characterized by a progressive obstruction to airflow and destruction of the lung parenchyma due to an inflammatory response and tissue damage, caused by chronic exposure to environmental factors in genetically susceptible individuals [1]. Inflammation is a term credited to Celsus. It was used as a metaphor because the dermal response to injury was reminiscent of fire, characterized by redness, heat, swelling, and pain. Virchow described a fifth change, loss of function (functio laesa). A sixth change, repair, could also be added, because a new growth occurs after tissue injury, just like after the fire occurs in forests, prairies and even cities in an attempt to preserve function and life [2]. When injury and inflammatory responses are abrogated, resorption of extracellular matrix protein occurs, promoting organ repair. When chronic injury persists, the unremitting activation of effector cells results in continuous deposition of extracellular matrix, progressive scarring and organ damage [3].
While it is true metaphor of Celsus remains available for academic purposes, semantics has oversimplified biological phenomena. The "inflammatory process" is incredibly complex, diverse and involves the participation of many cells and molecular products, interacting in complex combinations, activation of immune responses and pattern recognition receptors (PRRs). Briefly, the innate immune system is comprised of various anatomical barriers to infection, including physical barriers (bronchial epithelium and cilia), chemical barriers (bronchial mucus alkalinity), and biological barriers (oropharyngeal flora) [4]. Added to these barriers are soluble factors and phagocytic cells that form the first line of defense against pathogens. Soluble factors include the complement system, the reactant acute phase proteins and chemical messenger proteins called cytokines [5]. The complement system (a biochemical network of more than 30 proteins in plasma or cell surfaces) is a key component of innate immunity. The system develops responses that kill invading pathogens by direct lysis (cell disruption) or promote phagocytosis. These proteins also produce inflammatory responses which are an important part of innate immunity. The acute phase reactant proteins are a class of plasma proteins that are basic in inflammation. Cytokines secreted by immune cells in the early stages of inflammation stimulate the synthesis of acute phase reactant proteins by the liver [6]. Cytokines have a preponderant role in regulating the immune response; some cytokines interfere directly with pathogens. Interferons (IFNs) have antiviral activity [7]. Soluble factors are important to recruit phagocytic cells to local areas of infection such as monocytes, macrophages and neutrophils that engulf and digest invading microorganisms through a process called phagocytosis. In addition, neutrophils also form extracellular traps, which are chromatin networks containing antibacterial proteins that can trap and kill extracellular bacteria. These cells express the PRRs identifying the pathogen-associated molecular patterns (PAMPs) (diverse biochemical signatures) that are unique to pathogenic microorganisms but preserved through several families of pathogens. Immediate immune response is non-specific and has does not have "immunological memory", which means that the same response (same time, same intensity) must be orchestrated each time the system re-encounters the antigens.
Adaptive immunity (also called acquired immunity), a second line of defense against pathogens, takes several days, even weeks to develop to the fullest. However, adaptive immunity is much more complex because it involves an antigenic specificity and "immunological memory". Exposure to a specific antigen stimulates production of immune cells that target the pathogen for destruction [5]. “Immunological memory” means that the immune response to subsequent exposure of the same pathogen is faster and stronger because the antigens are "remembered." The primary mediators of this response are B and T lymphocytes. B lymphocytes produce antibodies which are specialized proteins that recognize and bind to foreign proteins or pathogens in order to neutralize or facilitate destruction by macrophages. The response mediated by antibodies is called humoral immunity. In contrast, cellular immunity is mediated by T lymphocytes, which develop in the thymus. Different subsets of T cells have different roles in immunity adaptive. For example, cytotoxic T (natural killer) cells directly attack and kill infected cells, while T helpers increase the response and help the function of other cells [4]. Regulatory T cells (also called suppressor T) suppress the immune response [5]. Memory T lymphocytes "remember" the antigens. In addition to its vital role in innate immunity, the complement system modulates the adaptive immune response and it is an example of interaction between the two immune systems [6]. Obviously, both response systems work together, to protect the body from infection and disease.
The PRRs are a family of sensors able to sense different exogenous microbial molecules as well as endogenous molecules which are released by the host tissue damage. The commitment of PRRs is a prerequisite for the initiation of inflammatory and immune responses to external aggression (for example, infection) and tissue injury that may be beneficial or harmful. The PRRs are germline-encoded, evolutionarily conserved molecules and consist of Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type lectin-like receptors (CLRs) and cytosolic DNA sensors. The PRRs are expressed in alveolar macrophages, epithelial lung cells, dendritic cells (DCs), endothelial cells and stromal cells as well as immune cells. PRRs not only recognize PAMPs, danger-associated molecular patterns (DAMPs), tumor-associated molecular patterns (TAMPs), but also large particles such as asbestos fibers, aluminum and silica crystals being critically involved in the pathogenesis of pneumoconiosis [8-11]. The cascade of events that generate inflammation is possibly the following: PRRs sense these molecules and produce signals leading to the production of part of the cells expressing them of inflammatory cytokines, IFNs and chemokines, which activate more cells around them (paracrine activity), macrophages and neutrophils (innate immune response) that, in turn, amplify the production and activity of more cytokines, capitalizing the reaction. The PRRs activate antigen presenting cells, such as macrophages and DCs, and with the participation of the major histocompatibility complex II (HLA-II), provides a mandatory signal to induce and activate T lymphocytes (Th1, Th2 and Th17) and production of antibodies (adaptive immune response) [12-15]. Therefore, both immune responses depend directly or indirectly on the PRRs recognition the molecules, and explain why these receptors play a key role in acute respiratory infection such as pneumonia, infectious exacerbation of COPD, inflammatory response to sterile tissue damage such as acute lung injury (ALI/ARDS) or disease from exposure to inorganic dust.
In both, inflammatory and autoimmune diseases, the condition originates through aberrant reactions of the adaptive immune response or the innate immune response. In chronic inflammatory diseases, neutrophils and other leukocytes are constitutively recruited by cytokines and chemokines, leading to tissue damage. In autoimmunity, the patient's immune system is activated against the proteins themselves. An autoimmune disease such as those caused by activation of T cells and / or B by auto-antigens can thus be defined in the absence of an infection or other discernible causes [16]. Is COPD an autoimmune disease? Discernible causes of COPD are the various types of stress generated by cigarette smoke and pollutants/biomass fuel. Infection is the leading cause of exacerbations of the entity. From this perspective it could be said that the initial mechanism that generates the inflammatory process in COPD is not autoimmune. From another angle, for a disease to be seen as autoimmune, it is necessary to fulfill Witebsky's postulates that say: direct evidence of transfer of T lymphocytes or antibodies that cause the disease, indirect evidence based on the reproduction of autoimmune disease in animals of experimentation and circumstantial evidence of clinical keys [17]. In the case of COPD, the three types of evidence have been established [18]. Even, some authors note that the chronic inflammatory process of the entity is immune [19]. Inflammation mediated by T cells in the lung, which persists for years after stopping smoking, has been identified as a key component of COPD and these findings suggest that COPD could be an autoimmune disease triggered by cigarette smoke [20]. This monograph therefore evaluates the evidence for and against the idea that COPD is an autoimmune disease.
Each inhalation of the cigarette smoke contains around 1017 reactive oxygen species (ROS) and more than 2000 xenobiotic compounds which initiate the inflammatory response in airways and lung parenchyma in COPD. This also occurs with exposure to biomass [21]. The inflammatory profile in normal smokers (without COPD) is very similar to that of patients with COPD, but less prominent. The concept that emerges is that of an amplified inflammatory response in COPD [22]. The amplified inflammatory response is associated with mucus production, proteolysis, fibrosis and resolution cycles. The process is possibly determined by genetic factors [23], latent viruses [24], oxidative stress [25] and alterations on the histone de-acetylase-2 (HDAC-2) activity [26]. The type of inflammation that occurs is mediated by the recruitment of different inflammatory cells and the production of distinct mediators (Figure 1).
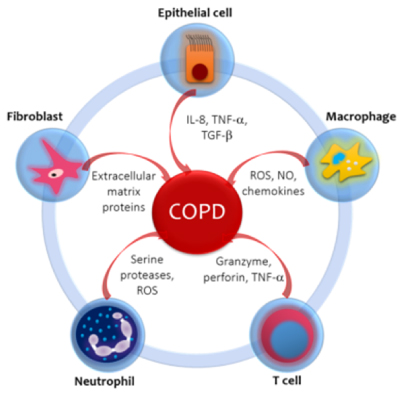
Figure 1. Inflammatory and immune cells and mediators involved in COPD. Cigarette smoke and other pollutants/biomass fuels are the initial noxa that stimulate epithelial cells and macrophages to release chemotactic factors that attract other inflammatory cells to the lungs. The distinct pattern of inflammation in COPD determines the structural consequences and response to therapy. The circular disposition is a simplified model, but it aims to emphasize the confluence and interplay between the related cells and mediators. IL-8: Interleukin-8. TNF-α: Tissue Necrosis Factor-α. TGF-β Transforming Growth Factor β. ROS: Reactive Oxygen Species. NO: Nitric Oxide.
Macrophages are increased in number and activity in the sputum and bronchoalveolar lavage (BAL) of patients with COPD; macrophages play a main role in orchestrating the inflammatory process. Stimulated by cigarette smoke and others irritants (biomass smoke), they release ROS, nitric oxide (NO) and chemokines that attract monocytes, T cells and neutrophils into the inflamed area [27]. T cells are CD8+ (suppressors / cytotoxic) and are located in the mucus secreting glands, central and peripheral airways and lung parenchyma. They release granzymes, perforins and tumor necrosis factor-α (TNF-α), which induces apoptosis of alveolar type I cells, favoring emphysema [26]. Neutrophils are increased in the sputum and BAL of patients with COPD. They are attracted by epithelial cells, macrophages and T cells through chemotactic factors such as interleukin 8 (IL-8), leukotriene B4 (LTB4) and a number of chemokines of the CXC family that act on specific receptors. Neutrophils release serine proteases like elastase, cathepsin G, proteinase-3, matrix metalloproteinase-12 (MMP-12) and ROS, which promote the mucus production and alveolar destruction [28].
It is controversial whether eosinophils are elevated in the sputum of stable COPD patients. Some recent studies claim that up to 40% of patients with COPD have airway eosinophilia and persistent peripheral eosinophilia with no history of asthma and that these patients are at greater risk of exacerbations than patients without eosinophilia [29,30]. 10% of COPD patients respond to inhaled glucocorticoids, these patients have a greater number of eosinophils in the airways and greater reversibility to bronchodilators. It has been suggested that these patients may have concomitantly asthma [31]. Recently, in a joint effort of GINA and GOLD the term ACOS was developed, as an overlap syndrome between asthma and COPD [32]. Epithelial cells produce TNF-α and IL-8 in response to inhaled bronchial irritants. They also generate transforming growth factor β (TGF-β) which can cause local fibrosis. Fibroblasts have increased activity and produce extracellular matrix proteins (ECM) in the small airways (obstructive bronchiolitis). Smooth muscle cells and endothelial cells are also involved in the inflammatory process [33].
The main proteases that elastin degrades (responsible for elastic recoil of the lung) are MMP9 and MMP12 and elastase and its activity is counterbalanced by inhibitors of metalloproteinases and α-1 AT (α-1 anti-trypsin). Cigarette smoke increases the release of proteases and decreases the production of inhibitors, clearly shifting the balance towards proteolysis. The cells that generate this displacement are the active cells in a chronic autoimmune inflammation directed against auto-antigens of the lung [18]. Human autoimmune disease is characterized by having in its profile activated T helper cells (Th1 and Th17) and their respective canonical cytokines (INF-γ, IL-17A and TNF) [34,35]. This cellular and immunological profile is essential for the human autoimmune disease model and has been demonstrated in emphysema and is like a signature of autoimmunity [36]. On the other hand, there is an increased prevalence of organ-specific autoimmune diseases in patients with COPD not associated with smoking [37]. They are usually older women, with lymphopenia, positive autoantibodies and airway inflammation. Patients with unexplained chronic cough and lymphocytic inflammation of the airway are 8 times more likely to have autoimmune disease and some will develop COPD not associated with smoking [38].
How smoke irritants trigger an immune innate response is unknown. The “danger hypothesis” suggests that epithelial injury (either an environmental or infectious insult) generates inflammatory mediators that activate macrophages and neutrophils that will produce oxidative stress, cell death, release of sequestered auto-antigens, modified proteins, damage mitochondrial and DNA release from apoptotic cells [39,40]. The adaptive immune response can recognize these antigens as foreign and trigger an immune reaction [41]. These, and other similar mechanisms have been implicated in rheumatoid arthritis, atherogenesis, multiple sclerosis, and systemic lupus erythematosus [42]. With environmental exposure, these auto-antigens can be released by necrosis and apoptosis of epithelial and endothelial cells and injury to the ECM [43]. Auto-antigens are not by themselves sufficient to develop an immune response or an autoimmune disease; however, PRRs (and particularly TLRs) are able to connect innate and adaptive immunity, increasing the pathogenic potential of these antigens [44]. If the chain of events does not progress, dendritic cells are not activated, innate immunity is minimized and the inflammatory process is contained, smokers may not develop COPD or develop early stages of the disease.
If the inflammatory cascade is not contained, immature antigen presenting cells (APCs), such as DC (myeloid and plasmocytoid), will alert the adaptive immune system to the presence of pathogens or tissue injury [45]. DCs mature when their TLRs bind ligands. Once mature, the DCs express high levels of the MHC proteins and the co-stimulatory molecules CD80 and CD86 direct them towards the local lymph nodes, where the DCs present the antigens to the T cells [46]. The ability of DC to present and stimulate T cells is what determines the progression of the disease. This ability is modulated by immunoregulatory mechanisms. If these mechanisms operate properly, the patient will not develop COPD. If the failure is slight, the early stages of the entity will arise. If the failure is severe, the process will continue until more advanced stages, which is what is detailed below.
The "naive" T cells will be attracted to the lung by means of chemokines that act on specific receptors. In COPD, basically CXCR3, CCR5 and CXCR6 are expressed in T lymphocytes (CD4+Th2 lymphocytes expressing CCR3 and CCR4 in bronchial asthma) (the letter R in the acronym indicates that it is a receptor). The ligands for these receptors are strongly expressed in structural cells of the airways and pulmonary arteries of patients with COPD but not in those of smoking patients without COPD. For example, for CXCR3 the ligand is CXCL10 (or interferon-inducible protein-10) [IP-10]) (the letter L in the acronym indicates that it is a ligand), or the CXCL9 (or monokine induced by interferon-γ [MIG]). The expression of these receptors and their ligands correlates with the severity of the disease [47]. The CD8+ cytotoxic T cell is the predominant cell in large pathways, small pathways and lung parenchyma in COPD, which perform a cytolytic attack releasing perforin, granulysin and granzimes A and B which are proteolytic enzymes that produce necrosis and apoptosis of epithelial cells and endothelial cells producing not only emphysema but also the release of more antigenic material, perpetuating autoimmunity and amplifying the process [18]. CD4+ T cells are also increased in the airways and parenchyma of patients with COPD. Its presence and activation in the lung (but not in blood), and the expression of STAT4 and interferon-γ suggest stimulation by antigens distributed through the lung. The effector function of CD4+ T cells is mediated by Th1 cytokines and is to promote transendothelial migration of inflammatory cells to the site of injury [48]. The increased function of dendritic cells, the genetic predisposition and the failure in tolerance or immune regulation catapult adaptive immunity and the severity of the disease towards advanced stages.
The NLR receptor family consists of 22 members in humans, and only some of them have been functionally characterized. Many of them, unlike the TLRs are fundamentally membranous, found in the cytosol. They all consist of a central NOD and a leucine rich repeats (LRRs) C-terminal which are those that mediate ligand binding possibly. They also have an N-terminal effector domain that allows subdividing the family on five subfamilies [49]. The proteins NLRP is a subgroup of NLRs which is composed of 14 proteins of which the NLRP1, NLRP3, NLRP6, NLRP7 and NLRP12 form multiprotein complexed called inflammasomes, which consist of one or two NLR proteins, adapter molecule ASC (apoptosis-associated protein speck-like containing a CARD) [PYCARD]) and procaspase-1 [50].The inflammasomes serve as a platform for autocatalytic activation of caspase-1 (by forming a zipper with procaspase-1) which critically regulates production of IL-1β, IL-18 and IL-33 processing pro-IL1β, pro-IL-18 and pro-IL-33 zymogens, and induce a form of cell death called pyroptosis [51]
The role of NLRP1 (was the first to be described) in lung disease in humans is unclear [52]. The NLRP3 inflammasome responds to a wide range of microbial and non-microbial molecules and has a more defined role in respiratory pathology. Chronic smoking activates NLRP3 as well as particles of matter and endogenous ATP acting as DAMPs [53,54]. Endogenous ROS product of NADPH bacterial oxidation and mitochondrial ROS activate NLRP3. It must be remembered that bacterial and viral infections are the main cause of exacerbations of COPD and source of endogenous ROS [55]. Patients with stable and exacerbated COPD have high levels of IL-1β (endogenous ATP, oxidative stress and infection), which keeps the inflammatory process active [56-58]. The other inflammasomes do not have enough studies to support their involvement in pulmonary pathology. Therapeutic manipulation of them already exists [59].
Studies of autoantibodies in COPD classify autoantigens into two types: undefined autoantigens (the majority) and defined antigens. The determination of autoantibodies is a proof of principle evidence. In the first group the antibodies are directed against cellular elements of lung or endothelial tissue, for example, IgG directed against airway epithelial cells and pulmonary artery endothelial cells [60,61]. The seconds are suspected to be functionally related to the entity. For example, autoantibodies against ECM such as those directed against elastin, collagen I, II and IV and aggrecan (another extracellular matrix protein), have been found elevated in patients with COPD, with higher titles than those of healthy controls, although not it's all jobs [62,63]. IgG, IgA, IgM antibodies directed against CK-18 and CK-19 (cytokeratins that are intracytoplasmic cyto-skeletal proteins of epithelial cells) they have also been reported in COPD [64]. Autoantibodies have also been described against immune cell molecules such as αβ-crystallin (a marker of activation of innate immunity), CD80 (co-stimulatory molecule of T lymphocyte activation), β2-microglobilin (component of MHC I) [65,66]. It is likely that the determination of the antibodies and their visualization depend not only on the autoantigens but also on the assay used.
A critical problem to sustain the preponderant role of autoimmunity in the pathogenesis of COPD is the inconsistent result of the studies. For example, the levels of anti-tissue antibodies correlate with the severity of emphysema (basically defined by the drop in DLCO), as well as the severity of COPD correlates with the levels of antibodies against CK-18 and CK-19 and CD80 [67]. This is not the case when correlating the extension of emphysema with anti-epithelial antibodies and with autoantibodies against elastin. The correlation between severity and activity of the antibodies is inversed.
In COPD there are several types of stress playing a pathogenic role. Oxidative stress can be exogenous or endogenous. Sources of exogenous ROS are chronic smoking, oxidant gases, ultrafine matter particles, environmental pollution nanoparticles, exhausted vapors and biomass fuels for cooking and heating homes (particularly in third world countries) [68,69]. Endogenous oxidants result mainly from mitochondrial respiration and inflammatory response to viruses and bacteria. Inflammatory stress is mediated by IL-1, TNF-α and INF-γ generating endogenous ROS. Other sources of intracellular ROS are the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase (XO) and hem-peroxidase, all of which are increased in BAL in patients with COPD. These enzymes catalyze the production of free radicals [70]. Others destructive oxidizing molecules are H2O2 (which acts as a substrate to generate additional oxidant molecules), ethane and isoprostanes (produced by direct oxidation of arachidonic acid). The superoxide anion and nitric oxide form the peroxynitrite that nitrosylates protein amino acids and enzymes, inactivating them. Therefore, ROS and reactive nitrogen species (RNS) are continuously produced at the cellular level by mitochondrial metabolism and by inflammatory cells in the case of hypoxic diseases as COPD. Endoplasmic reticulum (ER) oxidative stress can induce mitochondrial apoptosis and cell death. Normally, a fine cross-communication between ER and mitochondria prevents apoptosis of epithelial cells [71].
One type of neo-antigens (antigens expressed under specific pathophysiological conditions that are not ubiquitous in our body) are the carbonyl-modified proteins (CMP) generated by ROS. The reaction aldol in chemistry refers to the formation of carbon = carbon bonds (aldehydes and ketones). For example, oxidized lipids containing carbonyls derived from polyunsaturated fats (PUFAs) (lipid peroxidation in cell membranes produced by ROS) carbonyl certain amino acid residues of proteins such as the sulfhydryl groups of cysteine, γ-amino of lysine, and imidazole of histidine (the separation of the carbons by the covalent bond qualifies them as carbonyl adducts) [72]. Moderately carbonylated proteins are degraded by the cellular proteosomal system, but markedly carbonylated proteins tend to form high molecular weight aggregates that are resistant to degradation and accumulate as unfolded and damaged proteins, which inhibits the proteosomal activity. In other words, they are resistant to proteolysis [73].
Carbonylated proteins are highly immunogenic and lead to the formation of neo-antigens (CMP) in the form of carbonylated self-proteins, which leads to the formation of autoantibodies that are elevated not only in the serum of patients with COPD but also in the pulmonary epithelium and the endothelium of the pulmonary vessels [74]. Several proteins are converted to CMP. There are described autoantibodies for elastin, human serum albumin (HSA) and lung parenchyma, malonyldialdheyde protein, and acrolein-modified protein [72]. Circulating and lung antibodies are from the IgG1 isotype directed against carbonyl epitopes and could be the result of an aberrant immune response directed against putative antigens for which immune tolerance has been lost or never acquired. These antibodies bind complement (C3) and may contribute to the production of emphysema [75]. The presence of these reactive carbonyls and autoantibodies correlates inversely with FEV1 and the severity of the disease [76]. CMP are recognized by innate immunity through the PRRs, expressed in cells that identify antigens and from there the cascade of immune events described in the introduction is activated [77].
Mitochondria are complex cellular organelles assembled from proteins encoded by two distinct genomes: nuclear chromosomal DNA and the mitochondrial DNA (mtDNA). Despite the small size of the mtDNA (16.5 kb in humans), it encodes 13 potential oxidative phosphorylation subunits (OXPHOS) which interact with more than 70 units encoded by nuclear DNA; they concerted action is necessary to produce ATP, which is required for all active cellular processes [78]. Historically, the major role of mitochondria has been to catalyze the oxidation of metabolites for the production of ATP, via OXPHOS, involving the transference of high energy electrons derived from NADH and FADH2 to the four mitochondrial complexes, where they react with oxygen to form water. During this process, the protons are pumped through the inner mitochondrial membrane they are used by the FOF ATP synthase to generate ATP [79]. Additional critical functions are: regulation, proliferation, differentiation, cell death, redox and calcium homeostasis and recently, research has pointed to mitochondria as controlling the immune responses and determinants of immune cell phenotypes and their functions, including CD4+ T cells differentiation and CD8+ memory T cell formation [80]. Within the mitochondrial proteome there are key proteins such as mitochondrial antiviral signaling protein (MAVS), which is the first protein located in the mitochondria involved in the innate immune response and inflammatory response [81]. NOD-like receptor (NLRX1) is a member of the NLR family of PRRs that has a unique N-terminal domain (which explains the letter X in the acronym) and is the first, and only so far, of the PRRs located in the mitochondria and by therefore, it establishes a connection between mitochondrial function and innate immunity [82]. Apparently, the inhibition of inflammation and remodeling in COPD is performed by NLRX1 regulating MAVS [83].
In COPD, exposure to smoke reduces mitochondrial OXPHOS in airway smooth muscle cells, quadriceps, and external intercostal muscles, compromising oxidative function. Alteration of mitochondrial biogenesis may be associated with a significant reduction in body mass index and lower extremities muscle mass, a common phenomenon in COPD [84]. Loss of mtDNA has been associated with COPD [85]. Several independent studies have shown that NLRX1 protein expression and activity are suppressed by cigarette smoke in patients with COPD and this correlates with airflow obstruction. Basically, the idea is that chronic smoking alters NLRX1, inducing a mitochondrial dysfunction mediated by the NLRX1 / MAVS interaction. Chronic smoking, on inactivation of NLRX1, increases inflammatory activity (IL-18) favoring emphysematous destruction, mitochondrial ROS and proteases matrix metalloproteinase [85,86]. In this way, the three mechanisms (stress, autoimmunity and mitochondrial dysfunction) sometimes act in parallel, sometimes in series and often in concert, generating, amplifying and perpetuating the inflammatory phenomenon that underlies COPD.
Immune regulation, genetic susceptibility and environmental factors are what will define which smokers will develop or not an immune response to the material released by the lung damaged by the environmental noxa. And they will also be those that define the wide range of severity of the disease in response to similar amounts of exposure to smoke.
Existing pharmacologic treatments do not reduce the progression of COPD. Bronchodilators (which are the pharmacological basis of the therapy) only provide symptomatic relief [87]. Patients with COPD also have little response to glucocorticoids. This treatment has no effect on the progression of the disease or on mortality; a potential but slight decrease in exacerbations has been reported, although this finding has been questioned [88,89]. Antioxidant therapy in COPD is a logical approach and the topic has been recently reviewed [90], so the following item will refer to therapeutic alternatives that specifically impact autoimmunity in COPD.
For more than a decade it has been shown that cigarette smoke activates caspase-1 and caspase-11 and, in turn, these activate the IL-18 pro-peptide, which acts on its receptor (IL-18Rα). This process is a critical signal to induce inflammation and remodeling and it was suggested that blocking the IL-18 receptor could be a therapeutic alternative in respiratory pathology associated with chronic smoking [91]. It was also proposed that blocking the IL-1Rα receptor with Anakinra and the IL-1β receptor with Trap / rilonacept were possible options for pulmonary inflammation that leads to emphysema [92]. The sulfonylurea Glyburide (widely used drug for treatment of diabetes mellitus type 2) was shown to reduce NLRP3 inflammasome and was an option in the treatment of inflammatory lung diseases [93]. The results of the studies that aim to manipulate the immune loop of COPD have yielded dissimilar results. Monotherapy targeting Th1 and Th2 cytokines can be beneficial and deleterious. Monotherapy against IFN-γ may limit the progression of emphysema but may worsen vascular remodeling and airway remodeling, while monotherapy directed against IL-13 may limit the progression of pulmonary remodeling but accelerate the progression of emphysema [94]. Given that IL-18 has crucial antitumor activity as well as in activating host defenses and patients with COPD frequently they have infectious exacerbations and increased risk of developing lung cancer, it is vital to determine the safety and efficacy of novel therapies that have objective IL-18 in the lungs of patients with COPD [95]. The fundamental problem with COPD is that the underlying biological mechanisms in the pathogenesis are not fully known and by not having all the complete panoramic knowledge but fragmented, it becomes difficult the development of new drugs that are urgent to inhibit the chronic inflammation associated with COPD.
Some randomized clinical trials that target inflammasome-related effectors (i.e. IL-1α and IL-1β) have been performed on COPD patients considering moderate to severe stages. From 2007 to 2011 Novartis launched a phase 1/2 study (NCT00581945; Randomized, Double-blind, Placebo Controlled, Exploratory Study) to evaluate the safety, tolerability and efficacy of multiple doses of Canakinumab, a human anti-IL-1β monoclonal antibody, versus placebo when administered via intravenous infusion [96]. Pulmonary function in COPD patients was evaluated. Neither FEV1 nor FVC showed significant statistical differences among canakinumab-treated and placebo-treated COPD patients. Another study involving MED18968, a human IgG2 monoclonal antibody against IL-1R1, abled to bind both IL-1α and IL-1β, and injected intravenously (600 mg) and subcutaneously (300 mg) did not show any beneficial effect in COPD patients (GOLD stage II-IV) during exacerbations [97]. Similarly, MED12338, a human IgG1 monoclonal antibody that binds to IL-18, was studied for efficacy and safety in COPD and again, no statistical differences were observed between treated and placebo patients (clinical trial: NCT01322594). Moreover, based on the potential involvement of eATP and P2X7 in pathogenesis of COPD, recently the effect of AZD9056, a P2XT antagonist which is able of bind the human P2XT receptor with high selectivity and specificity, was studied in Phase 2 clinical trials in patients with moderate to severe COPD, but again the pulmonary parameters studied were not significantly altered by the antagonist [98].
The use of recent biological treatments comes originally from studies in populations of patients with difficult-to-control asthma, also labeled as severe asthma [99,100]. These patients may have basically two immunological profiles. The eosinophilic profile, characteristic of airway inflammation type 2, characterized by the presence of cytokines originally recognized as being produced by type helper T (Th2) cells. Th2 cytokines are IL-4, IL-5 and IL-13. These cytokines are also produced by innate lymphoid cells (which do not express B or T cell receptors) in response to infectious agents, pollutants and other "non-allergic" stimuli. These patients persistently maintain this inflammatory profile despite high doses of inhaled glucocorticoids [101-104]. The other profile is neutrophilic inflammation (defined as exceeding 40-60% neutrophils) in induced sputum samples.
Many of the recent approved biological interventions target type 2- inflammation. IL-5 plays a central role in promoting eosinophilic inflammation. Anti-IL-5 monoclonal antibodies are now available for the treatment of patients with severe eosinophilic asthma and recurrent exacerbations. Mepolizumab and reslizumab, both of which bind to IL-5, have been approved by several regulatory agencies in the US and EU. Benralizumab, which binds to the IL-5 receptor, producing eosinophilic apoptosis, is nearing Food and Drug Administration (FDA) approval [105]. Blockade of IL-13 has the potential to alter airway inflammation and smooth-muscle reactivity, but one of the two anti-IL-13 monoclonal antibodies, lebrikizumab, failed to provide consistent improvement in patients with type 2. inflammation. The other, tralokinumab, continues in development [106]. Dupilumab is a full human monoclonal antibody to the alpha subunit of the IL-4 receptor that blocks both (IL-4 and IL-13) signaling [107]. Based on this platform of immune knowledge and this experience with eosinophilic asthma, studies have been designed using biologic interventions in patients with COPD.
The use of mepolizumab in patients with COPD and eosinophilic phenotype (2% or more [150-200 eosinophils / mm3] in peripheral blood) and who already received triple therapy (including inhaled glucocorticoids) has resulted in a reduction in the rate of moderate to severe exacerbations of COPD when compared to placebo and also in a longer time until the first exacerbation, and the extension of this effect was related to the blood eosinophilia count [108]. The stratification of COPD phenotypes could make it possible to clearly define the subgroups of patients that could benefit from this type of therapy. For example, Xia and colleagues suggest that some of the patients in the aforementioned work protocol qualified as ACO (overlap between asthma and COPD) and, therefore, that the biological could be of benefit in ACO [109]. This indication of mepolizumab to mitigate severe eosinophilic asthma in patients who also show changes in COPD has already been reported [110]. Similar results have been reported in patients with ACO in studies with dupilumab (anti-IL-4) and omalizumab (anti-IgE) [111,112].
Some authors consider that blood eosinophilia is inferior to sputum eosinophilia as a biomarker in these patients. The presence of allergy and atopy, the disparity of specificity and sensitivity between blood eosinophil count and sputum, and the fact that some previous studies do not agree that eosinophilia is a generalized biomarker to define COPD phenotypes, support this consideration [113]. Other researchers do not find a robust association between blood eosinophilia and the occurrence of exacerbations. One of the fundamental problems with adding biological therapies in these patients is the high price. And, if that is a limitation in high-income countries, with much more reason in third world countries with meager health budgets [114,115].
It is obvious that much remains to be defined in this immunological approach to COPD. Since the inflammatory and immune process is complex, probably unraveling the start of the cascade of events is the best way to understand it. As ROS at the molecular level triggers the innate immune response should be a research priority. This could allow finding markers of the initial lesion that had early diagnostic value and also implement safer therapeutic strategies.
Another important area of research is the immunoregulatory mechanisms, because if they were known they could be manipulated pharmacologically or epigenetically.
It is also vital to better define the autoantigens of epithelial and endothelial cells. For example, identify the auto-antigens of endothelial cells that may be involved in the genesis or in the perpetuation of pulmonary hypertension secondary to COPD.
Another important aspect is to try to standardize the immunological assays for the detection and quantification of antibodies.
Improving monoclonal antibodies in terms of efficacy and safety is another priority.
Mitochondria offer a research objective not only for diagnosis but also for the treatment of pulmonary diseases. Transfer of mitochondria from mesenchymal stem cells from bone marrow to injured epithelial cells could be beneficial in COPD [116]. Contact is important for the transmission of calcium signals from the ER to the mitochondria. This is done by proteins. Therefore, designing strategies that refine and perpetuate transmission could improve the function and survival of mitochondria [117].
The mortality of COPD, worldwide, continues to rise.
The treatment recommended by international guidelines does not significantly impact lung function or the natural evolution of the disease. This may be due to the fact that cyclic infection and autoimmunity play a role in the development, perpetuation and chronicity of the inflammatory process.
Despite the large amount of evidence that autoimmunity has a predominant pathogenic role, the cause-effect relationship is not completely proven, so that COPD can´t be defined as a primary autoimmune disease. What does appear is that the autoimmune phenomenon can be triggered by chronic exposure to smoke.
The autoimmune phenomenon in COPD is well documented and seems to have a pathogenic role and not only be an epiphenomenon.
Defining the axis of stress, autoimmunity and mitochondrial dysfunction better could generate diagnostic and therapeutic benefits.
Therefore, future and extensive investigations are required in the entity to unravel and understand the complex signals that initiate, develop and perpetuate the process.
This is vital to implement early diagnostic tools and new therapeutic tools.
No.
No.
This work was only carried out by the author. Author AA contributed in the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
- Capistrano SJ, van Reyk D, Chen H (2017) Evidence of Biomass Smoke Exposure as a Causative Factor for the Development of COPD. Toxics 5. [Crossref]
- Rennard SI (2004) Antiinflammatory therapies other than corticosteroids. Proc Am Thorac Soc 1: 282-287. [Crossref]
- Rockey DC, Bell PD, Hill JA (2015) Fibrosis--a common pathway to organ injury and failure. N Engl J Med 372: 1138-1149. [Crossref]
- Parkin J, Cohen B (2001) An overview of the immune system. Lancet 357: 1777-1789. [Crossref]
- Delves PJ, Roitt IM (2000) The immune system. First of two parts. N Engl J Med 343: 37-49. [Crossref]
- Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20: 34-50. [Crossref]
- Mak TW, Saunders ME (2004). Innate immunity. The immune response: basic and clinical principles. Amsterdam: Elsevier Academy Press p. 79-92.
- Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S, et al. (2008). Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3: 352-363.
- Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27: 229-265. [Crossref]
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, et al. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674-677. [Crossref]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, et al. (2008). Silica crystals and aluminium salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9 : 847-856.
- Fritz JH, Le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, et al. (2007). Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 26: 445-459.
- van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller EJ, Hommes DW, et al. (2007) Stimulation of the intracellular bacterial sensors NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 27: 660-669.
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, et al. (2008) TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451: 725-729.
- Meng G, Zhang F, Fuss I, Kitani A, Strober W (2009) A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity 30: 860-874.
- Zhernakova A, van Diemen CC, Wijmenga C (2009) Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet 10: 43-55. [Crossref]
- Rose NR, Bona C (1993) Defining criteria for autoimmune diseases (Witebsky’ postulates revisited). Immunol Today 14: 426-430.
- Cosio MG, Saetta M, Agusti A (2009) Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 360: 2445-2454. [Crossref]
- Hemminki K, Liu X, Ji J, Sundquist K, Sundquist J (2011) Subsequent COPD and lung cancer in patients with autoimmune disease. Eur Respir J 37: 463-465. [Crossref]
- Cosio MG (2004) Autoimmunity, T-cells and STAT-4 in the pathogenesis of chronic obstructive pulmonary disease. Eur Respir J 24: 3-5. [Crossref]
- Salvi S, Barnes PJ (2010) Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest 138: 3-6. [Crossref]
- Gold Reports (2018) Global initiative for the diagnosis, management and prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease (GOLD).
- Castaldi PJ, Cho MH, Langerman F, Moran S, Tarragona N, et al. (2010). The COPD genetic association compendium: a comprehensive genetic association. Hum Mol Genet 19: 526-534.
- Meshi B, Vitalis TZ, Ionescu D, Elliott WM, Liu C, et al. (2002) Emphysematous lung destruction by cigarette smoke. The effects of latent adenoviral infection on the lung inflammatory response. Am J Respir Cell Mol Biol 26: 52-57. [Crossref]
- Rahman I (2005) Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys 43: 167-188.
- Barnes PJ, Shapiro SD, Pauwels RA (2003) Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 22: 872-888.
- Alvarado A, Arce I (2015) Molecular biology of chronic obstructive pulmonary disease from the bases to the therapeutic decision: a review. Br J Med Medic Res 10: 1-14.
- Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, et al. (2009) MMP12, lung function, and COPD in high-risk populations. N Engl J Med 361: 2599-2608. [Crossref]
- Singh D, Kolsum U, Brightling CE, Locanfore M, Augusti A, et al. (2014) Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J 44: 1697-1700.
- Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG (2016) Blood eosinophils and exacerbations in chronic obstructive pulmonary disease-the Copenhagen general population study. Am J Respir Crit Care Med 193: 965-974.
- Barnes PJ (2008) Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183-192. [Crossref]
- (ACOS) (2018) Diagnosis of Asthma, COPD and asthma-COPD overlap syndrome. A joint project of GINA and GOLD.
2021 Copyright OAT. All rights reserv
- Hansel TT, Barnes PJ (2003) An atlas of chronic obstructive pulmonary disease: COPD- A resource for reference, teaching and lecturing. (1st Edn) London: Taylor & Francis.
- Palmer MT, Weaver CT (2010) Autoimmunity: increasing suspects in the CD4+ T cell lineup. Nat Immunol 11: 36-40. [Crossref]
- Shan M, Yuan X, Song L, Robert L, Zarinkamar N, et al. (2012) Cigarette smoke induction of osteopontin (SPP1) mediates Th17 inflammation in human and experimental emphysema. Sci Transl Med 4: 117ra9.
- Kheradmand F, Shan M, Chuang X, Corry DB (2012) Autoimmunity in chronic obstructive pulmonary disease: clinical and experimental evidence. Expert Rev Clin Immunol 8: 285-292.
- Birring SS, Brigtling CE, Bradding P, Entwisle JJ, Vara DD, et al (2002) Clinical, radiological and induced sputum features of chronic obstructive pulmonary disease in nonsmokers: a descriptive study. Am J Respir Crit Care Med 166: 1078-1083.
- Birring SS, Pavord ID (2011) COPD: an autoimmune disease? Eur Respir J 38: 484. [Crossref]
- Matzinger P (2002) The danger model: a renewed sense of self. Science 296: 301-305. [Crossref]
- Krieg AM, Vollmer J (2007) Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev 220: 251-269. [Crossref]
- Marrack P, Kappler J, Kotzin BL (2001) Autoimmune disease: why and where it occurs. Nat Med 7: 899-905. [Crossref]
- Rose N, Afanasyeva M (2003) Autoimmunity: busting the atherosclerotic plaque. Nat Med 9: 641-642. [Crossref]
- Steinman L. (2006). State of the art: easy pieces, interconnection between tissue injury, intermediary metabolism, autoimmunity, and chronic degeneration. Proc Am Thorac Soc 3: 484-486.
- Pasare C, Medzhitov R (2004) Toll-like receptors and acquired immunity. Semin Immunol 16: 23-26. [Crossref]
- Hemmi H, Akira S (2005) TLR signalling and the function of dendritic cells. Chem Immunol Allergy 86: 120-135 [Crossref]
- Lambrecht BN, Prins JB, Hoogsteden HC (2001) Lung dendritic cells and host immunity to infection. Eur Respir J 18: 692-704. [Crossref]
- Freeman CM, Curtis JL, Chensue SW (2007) CC chemokine receptor and CXC chemokine receptor 6 expression by lung CD8+ cells correlates con chronic obstructive pulmonary disease severity. Am J Pathol 171: 767-776.
- Shapiro SD (2001) End stage chronic obstructive pulmonary disease: the cigarette is burned out but inflammation rages on. Am J Respir Crit Care Med 164: 339-340.
- Chen G, Shaw MH, Kim YG, Nuñez G (2009) NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 4: 365-398. [Crossref]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140: 821-832. [Crossref]
- Chaput C, Sander LE, Suttorp N, Opitz B (2013) NOD-Like Receptors in Lung Diseases. Front Immunol 4: 393. [Crossref]
- Kummer JA, Broekhuizen R, Evertt H, Agostini L, Kuisk L, et al. (2007). Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissue suggesting a site-specific role in the inflammation response. J Histochem Cytochem 55: 443-452.
- Sayan M, Mossman BT (2016) The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol 13: 51. [Crossref]
- De Falco G, Terlizzi M, Sirignano M, Commodo M, D’Anna A, et al. (2017). Human peripheral blood mononuclear cells (PBMCs) from smokers release higher levels of IL-1-like cytokines after exposure to combustion-generated ultrafine particles. Sci Rep 7: 43016.
- Alvarado A, Arce I (2016) Immune recognition in lung diseases: basic research and clinical application. Clin Infect Immun 1: 31-40.
- De Nardo D, De Nardo CM, Latz E (2014) New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol 184: 42-54. [Crossref]
- Rovina N, Koutsoukou A, Koulouris NG (2013) Inflammation and immune response in COPD: where do we stand? Mediators Inflamm 2013: 413735. [Crossref]
- Faner R, Sobradillo P, Noguera A, Gomez C, Cruz T, et al. (2016) The inflammasome pathway in stable COPD and acute exacerbations. ERJ Open Res 2. [Crossref]
- Colarusso C, Terlizzi M, Molino A, Pinto A, Sorrentino R (2017). Role of inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget 8: 81813-81824.
- Nunez B, Sauleda J, Anto JM, Julia MR, Orozco M, et al. (2011).Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183: 1025-1031.
- Karayama M, Inui N, Suda T, Nakamura Y, Nakamura H, et al. (2010) Antiendothelial Cell Antibodies in Patients With COPD. Chest 138: 1303-1308. [Crossref]
- Brandsma CA, Kerstjens HA, Geerlings M, Kerkhof M, Hylkema MN, et al. (2011) The search for autoantibodies against elastin, collagen and decorin in COPD. Eur Respir J 37: 1289-1292. [Crossref]
- Daffa NI, Tighe PJ, Corne JM, Fairclough LC, Todd I (2015) Natural and disease-specific autoantibodies in chronic obstructive pulmonary disease. Clin Exp Immunol 180: 155-163.
- Xion Y, Gao S, Luo G, Cheng G, Jiang R, et al (2017). Increased circulating antiantibodies levels of IgG, IgA, IgM against cytokeratin 18 y cytokeratin 19 in chronic obstructive pulmonary disease. Arch Med Res 48: 79-87.
- Cherneva RV, Georgiev OB, Petrova DS, Trifonova NL, Stamenova M, et al (2012). The role of small heath-shock protein alphaß-crystalline (HspB5) in COPD pathogenesis. Int J Chron Obstruct Pulmon Dis 7:633-640.
- Luo XM, Liu XY, Tang JH, Yang W, Ni ZH, et al. (2016) Autoantibodies against CD80 in patients with COPD. Clin Transl Immunology 5: e103. [Crossref]
- Wen L, Krauss-Etschmann S, Petersen F, Yu X1, et al. (2018) Autoantibodies in Chronic Obstructive Pulmonary Disease. Front Immunol 9: 66. [Crossref]
- Salvi SS, Barnes PJ (2009) Chronic obstructive pulmonary disease in non-smokers. Lancet 374: 733-743. [Crossref]
- Kokasl H, Saygi A, Sariman N, Alici E, Yurtlu S, et al. (2013) Evaluation of clinical and functional parameters in female subjects with biomass smoke exposure. Respir Care 58: 424-430. [Crossref]
- Aaron SD, Angel JB, Lunau M, Wright K, Fex C, et al. (2001). Granulocyte inflammatory markers and airway infection during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 349-355.
- Obeng EA, Boise LH (2005) Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem 280: 29578-29587.
- Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, et al. (2011). Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184: 796-802.
- Dalle-Donne J, Aldini G, Carini M, Colombo R, Rossi R, et al. (2006). Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med 10: 389-406.
- Eggleton P1, Haigh R, Winyard PG (2008) Consequence of neo-antigenicity of the 'altered self'. Rheumatology (Oxford) 47: 567-571. [Crossref]
- Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, et al (2008). Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177: 156-173.
- Barreiro E, de la Puente B, Minguella J, Corominas JM, Serrano S, et al. (2005). Oxidative stress and respiratory muscle dysfunction in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 171:1116-1124.
- Allison ME, Fearon DT (2000) Enhanced immunogenicity of aldehyde-bearing antigens: a possible link between innate and adaptive immunity. Eur J Immunol 30: 2881-2887.
- Chinnery PF, Zeviani M (2016) Mitochondrial Matchmaking. N Engl J Med 375: 1894-1896. [Crossref]
- Semenza GL1 (2011) Oxygen sensing, homeostasis, and disease. N Engl J Med 365: 537-547. [Crossref]
- Bialas AJ, Sitarek P, Milkowska-Dymanowska J, Protrowski WJ, Gorski P. (2016) The role of mitochondrial and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cel Longev 2016: 7808576.
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122: 669-682. [Crossref]
- Arnoult D, Soares F, Tattoli I, Girardin SE (2011) Mitochondria in innate immunity. EMBO Rep 12: 901-910. [Crossref]
- Kang MJ, Yoon CM, Kim BH, Lee CM, Zhou Y, et al. (2015) Suppression of NLRX1 in chronic obstructive pulmonary disease. J Clin Invest 125: 2458-2462. [Crossref]
- Rabinovich RA, Vilaro J (2010) Structural and functional changes of peripheral muscles in COPD patients. Curr Opin Pulm Med 162: 122-133.
- Mizumara K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, et al. (2014) Mitophagy-dependent necroptosis contributes in the pathogenesis of COPD. J Clin Invest 124: 3987-4003.
- Kang MJ, Shadel GS (2016) A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis 79: 207-213.
- Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, et al. (2008) A 4-year trial of tiotropium (UPLIFT) on patients with chronic obstructive pulmonary disease. N Engl J Med 359: 1543-1554.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD (2008) Methodological issues in therapeutic trials of COPD. Eur Respir J 31: 927-933. [Crossref]
- Bourbeau J, Christodoulopoulos P, Maltais F, Yamauchi Y, Olivenstein R, et al. (2007). Effect of salmeterol/fluticasone propionate on airway inflammation in COPD: a randomized controlled trial. Thorax 62: 938-943.
- Alvarado A (2017) Antioxidants in respiratory diseases: Basic science research and therapeutic alternatives. Clin Res Trials 3: 1-11.
- Kang MJ1, Homer RJ, Gallo A, Lee CG, Crothers KA, et al. (2007) IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J Immunol 178: 1948-1959. [Crossref]
- Couillin I, Vasseur V, Charron S, Gasse P, Tavernier M, et al. (2009) IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J Immunol 183: 8195-8202. [Crossref]
- Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, et al. (2009) Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187: 61-70. [Crossref]
- Nakajima T, Owen CA (2012) Interleukin-18: the master regulator driving destructive and remodeling processes in the lungs of patients with chronic obstructive pulmonary disease? Am J Respir Crit Care Med 185: 1137-1139. [Crossref]
- Koshiol J, Rotunno M, Consonni D, Pesatori AC, De MS, et al (2009). Chronic obstructive pulmonary disease and altered risk of lung cancer in a population-based case-control study. PLoSONE 4: e7380.
- Rogliani P, Calzetta L, Ora J, Matera MG (2015) Canakinumab for the treatment of chronic obstructive pulmonary disease. Pulm Pharmacol Ther 31: 15-27. [Crossref]
- Calverley P, Sethi S, Dawson M, Ward C, Newbold P, et al. (2015). A phase 2 study of MED18968, an interleukin-1 receptor (IL-1R) monoclonal antibody, in adults with moderate-to-very severe chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 191: A3964.
- https://neats.nih.gov/files/AZD9056.pdf
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, et al. (2014) International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 43: 343-373. [Crossref]
- Hekking PP, Wener RR2, Amelink M2, Zwinderman AH3, Bouvy ML4, et al. (2015) The prevalence of severe refractory asthma. J Allergy Clin Immunol 135: 896-902. [Crossref]
- Wenzel SE (2012) Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18: 716-725. [Crossref]
- Brusselle GG, Maes T, Bracke KR (2013) Eosinophils in the spotlight: Eosinophilic airway inflammation in nonallergic asthma. Nat Med 19: 977-979. [Crossref]
- Wu W, Bleccker E, Moore W, Buse WW, Castro M, et al. (2014). Unsupervised phenotyping of Severe Asthma Research Program using expanded lung data. J Allergy Clin Immunol 133: 1280-1288.
- Lambrecht BN, Hammad H (2015) The immunology of asthma. Nat Immunol 16: 45-56. [Crossref]
- Pavord ID, Korn S, Howarth P, Bleccker ER, Keene ON, et al (2012). Mepolizumab for severe eosinophilic asthma (DREAM): a multicenter, double-blind, placebo-controlled trial. Lancet 380: 651-659.
- Hanania NA, Noonan M, Corren J, Korenblat P, Zheng Y, et al. (2015) Lebrikizumab in moderate-to-severe asthma: pooled data from two randomised placebo-controlled studies. Thorax 70: 748-756. [Crossref]
- Wenzel S, Castro M, Corren J, Maspero J, Wang L, et al (2016). Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus long-acting ß2 agonist: a randomized, double-placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 388: 31-44.
- Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Korn S, et al. (2017). Mepolizumab for eosinophilic chronic obstructive disease. N Engl J Med 377: 1613-1616.
- Xia Y, Li W, Shen H (2018) Mepolizumab for Eosinophilic COPD. N Engl J Med 378: 680-681. [Crossref]
- Yarcey S, Albers FC, Gunsoy N, Harris S, Keene O (2017) Effect of mepolizumab on exacerbations in asthma patients with features common in COPD. Am J Respir Crit Care Med. 195: A4683-A4683.
- Wenzel S, Jayawardena S, Graham NM, Pirozzi G, Teper A, et al. (2016). Severe asthma and asthma-chronic obstructive pulmonary disease syndrome-authors’reply. Lancet 388: 2742-2742.
- Maltby S, Gibson PG, Powell H, McDonald VM. (2017). Omalizumab treatment response in a population with severe allergic asthma and overlapping COPD. Chest 151:78-89.
- Shahlavi-Monavvar P, Mobasher-Jannat A (2018). Mepolizumab for eosinophilic COPD. To the editor. N Engl J Med 378: 680-683.
- Keene JD, Jacobson S, Kechris K, Kinney GL, Foreman MG, et al. (2017) Biomarkers Predictive of Exacerbations in the SPIROMICS and COPDGene Cohorts. Am J Respir Crit Care Med 195: 473-481. [Crossref]
- Alvarado A (2017) Dual bronchodilator therapy: a review. Clin Res Trials 3: 1-12.
- Kang MJ, Shadel GS (2016) A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis (Seoul) 79: 207-213.
- Mishra P, Chan DC (2014) Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15: 634-646. [Crossref]