Abstract
Introduction: Although platelets have a special relevance in the formation and maintenance of thrombus that occurs after plaque rupture, a consensus has emerged that inflammation also plays a decisive role in the pathophysiology of these acute thrombotic events.
Antiplatelet treatment is the “gold standard” for ACS. However, some patients develop recurrent CV events besides the antiplatelet treatment. Different hypothesis tried to explain these results, including genetic differences. But recently the relationship between platelet turn-over, a hyper-inflammatory status and prothrombotic risk are emerging.
Areas covered: Inflammatory cytokines accelerate platelet daily turn-over and the release of an increased number of fresh immature platelets may not be effectively inhibited by the once-a-day administration of antiplatelet drugs. Young platelets appear to participate most actively in the thrombotic process.
Expert opinion: Combination of antiplatelet drugs, more potent antiplatelet and higher doses proved to be not enough to reduce thrombotic events and they have increased the bleeding risk. Fragmented low doses of antiplatelet drugs during the day could be an alternative to evaluate according to some evidence.
Key words
antiplatelet, dosing, fragmented, turn-over, twice
Introduction
Platelets are small (1-3 um), circulating anucleated blood cells that play a key role in the development of acute coronary syndromes and contribute to cerebrovascular events [1]. The usual mechanism responsible for the sudden transition from a stable, often clinically silent, atherosclerotic disease to a symptomatic life-threatening condition is the erosion or disruption of an atherosclerotic lesion that will trigger the acute formation of a thrombus [1]. Thrombosis is the result of a complex set of interactions between platelets, coagulation factors, and the vessel wall.
Although platelets have a special relevance in the formation and maintenance of thrombus that occurs after plaque rupture, a consensus has emerged that inflammation also plays a decisive role in the pathophysiology of these acute thrombotic events [2]. Inflammation is considered essential to the initiation, development, and progression of atherosclerosis [3].
Inflammatory processes in the atherosclerotic artery (leading to macrophage recruitment) are associated with increased blood levels of inflammatory cytokines and other acute-phase reactants. The presence of several inflammatory biomarkers, most notably high-sensitivity C reactive protein (CRP), are considered predictors for coronary events [4] and have demonstrated prognostic value in acute coronary syndromes [5].
Moreover, multiple studies have shown that a high white blood cells (WBC) count is associated with increased mortality rates among patients presenting acute coronary syndromes (ACS) [6], in acute myocardial infarction (AMI) [7], in patients who undergo percutaneous coronary interventions [8] (PCI) and in patients with cerebral ischemia or stroke[9].
Interleukin-6, a crucial cytokine for leukocyte and endothelial cell activation and the hepatic promoter for CRP generation, [10] in addition to more than 40 inflammatory biomarkers have been found to be useful for detecting coronary artery disease and prognosis [3].
Antiplatelet treatment is the “gold standard” for ACS. However, some patients develop recurrent CV events besides the antiplatelet treatment [11-13]. Different hypothesis tried to explain this results, including genetic differences [14-17] but more recently the relationship between platelet turn-over and an hyper-inflammatory status are emerging [18-21].
Cross– talk between platelets, endothelium and leukocytes
Interactions between an inflamed/activated endothelium and platelets are mediated by the expression of adhesive proteins on the endothelium (e.g. P-selectin, ICAM, VCAM) that facilitate platelets and endothelium interaction leading to platelet secretion. These events perpetuate the inflammatory interactions between platelets, endothelial cells, and leukocytes [22].
Activated macrophages, T cells and mast cells, release several types of molecules (inflammatory cytokines, proteases, coagulation factors, radicals, and vasoactive molecules) facilitating plaque rupture and subsequent acute thrombus formation. Two types of proteases have been implicated as key players in plaque activation: matrix-metallo-proteinases (MMPs) and cysteine-proteases. These enzymes, by digesting the extracellular matrix, contribute to plaque destabilization and ACS manifestations.
However, the most important relationship between platelets and inflammation might be the accelerated platelet turn-over that occurs during an ACS. In healthy subjects, it is estimated that the total circulating platelets are replace within 7-10 days. Several pathological conditions have been associated with an accelerated platelet turn- over, being diabetes, smoking, extensive atherosclerosis and inflammatory conditions among them [23]. Inflammatory disorders are commonly associated with thrombocytosis. The relationship with inflammatory states could be mediated via IL6-induced megakaryocyte proliferation through thrombopoyetin [24]. Of interests, presence of increased inflammatory biomarkers has been associated to sub-optimal response to antiplatelet agents.
The “fiction of the perpetually platelet inactivation”
For long time, it has been considered that a daily aspirin (ASA) dose was enough to inhibit platelet aggregation [25]. As platelets are anucleated cells, and ASA inhibits cyclo-oxigenase (COX) in an irreversibility manner, it has been interpreted that the effect lasts through the life-span of the platelet and the recovery of platelet function requires the regeneration of new platelets form megakaryocytic cells [26].
Studies in healthy subjects have indicated that platelet thromboxane (TX) synthesis needs to be blocked to 90% to achieve efficient platelet inhibition. Thus, while the daily platelet turn-over is around 10%, the blockade of the COX-1 enzyme by once daily ASA seems to be enough to offer an adequate platelet aggregation inhibition. However, increasing attention has been given to inter-individual variability and/or non-response to different antiplatelet drugs.
Henry P, et al. demonstrated in a population of atherosclerotic patients chronically treated with aspirin that daily dose of aspirin cannot effectively inhibit the platelet thromboxane pathway for the 24 hours [27]. This reduced response has been related to the presence of high levels of inflammatory markers [24] and increased platelet turnover [28].
Platelet count and mass are highly modulated by thrombopoietin, which is activated by IL-6. High levels of thrombopoietin have been reported in inflammatory states [29] and within days of an acute myocardial infarction [30]. It leads to reactive thrombocytosis and elevated platelet count resulting in more recurrent events. High platelet numbers have been independently associated with higher mortality and MACE in ACS patients [21,31].
Two possible explanations can be hypothesized:
- Inflammatory cytokines (e.g. IL-6) capable of stimulating thompoietin release will also accelerate platelet daily turn-over above 10-20%. The release of an increased number of fresh platelets may not be effectively inhibited by the once-a-day administration of antiplatelet drugs. Therefore, the non-inhibited platelets may allow the recovery of blood reactivity given the number of naïve platelets that have no contact with the antiplatelet drug, depending on the half-life of the specific antiplatelet agent (the “platelet quantity hypothesis”). Figure 1.
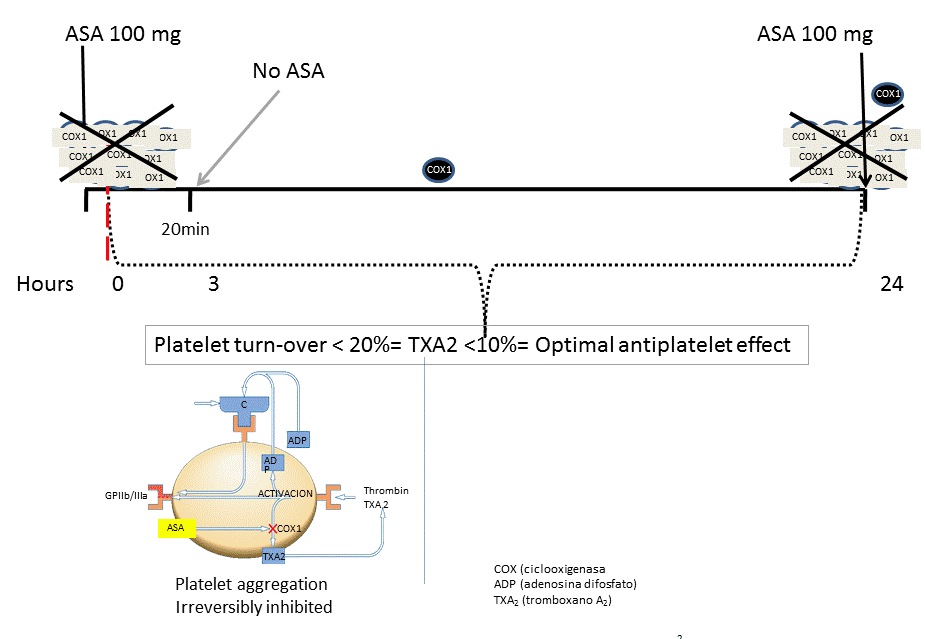
Figure 1a. Effect of ASA with normal platelet turnover.
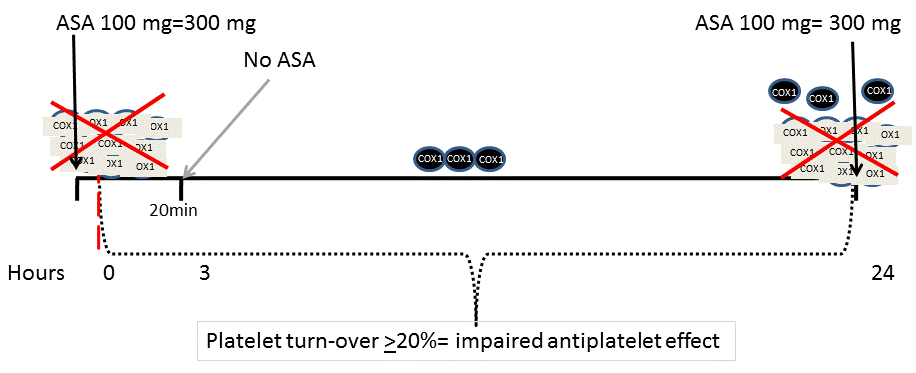
Figure 1b. Increased platelet turn-over and the once-a-day administration of antiplatelet drugs. The release of an increased number of fresh platelets may not be effectively inhibited by once-a-day administration of antiplatelet drugs.
Although the precise mechanism behind the relationship between elevated platelet count and adverse outcomes in patients with ACS is unknown, a possible explanation is that a higher platelet count directly may release immune mediators (including P-selectin, intercellular adhesion molecule 2, toll-like receptors, CD40 and CD40 ligand, etc) significantly favoring atherosclerosis progression and contributing to the formation and stabilization [32].
- The population of circulating platelets is not homogeneous [33], immature platelets comprise the youngest component of the circulating platelet pool, and appear to participate most actively in the thrombotic process [34]. They contain measurable amounts of cytosolic messenger ribonucleic acid (mRNA) that is translationally active [35].
There is concordance between the level of circulating young platelets and the proportion of subjects who are hyporesponsive to aspirin or clopidogrel, indicating that the presence of young platelets may attenuate the inhibitory effects of these agents [28,33,36] (the “platelet quality hypothesis”).
Although this mRNA was at one time considered inconsequential, a growing body of evidence indicates the presence of spliceosome complexes in these platelets and that activation-dependent splicing and translation of pre-mRNA results in the de novo synthesis of prothrombotic and proinflammatory proteins [28,35]. Ibrahim et al demonstrated that the number of immature platelets could be considered a biomarker for MACE risk stratification in patients with coronary artery disease [37,38].
Observations supporting the hypothesis and strategies trying to sort the problem
Platelet number and immature platelets, probably due to their higher reactivity [28], have been shown to be of prognostic value in different clinical scenarios such as ST-Elevation Myocardial Infarction [31], non STEMI, unstable angina [21] and stable coronary artery disease [36]. Moreover, significantly elevated levels of platelet–monocyte aggregates have been found in patients with acute myocardial Infarction and unstable angina; observation again compatible with a hyper-reactive state in both cells [20].
These observations strongly suggest the notion that identification and quantification of platelet–monocyte binding in patients with chest pain may provide key early in vivo evidence of vascular injury responses and offer opportunities for novel therapeutic intervention strategies in the treatment of ACS [15].
On the other hand, recent genetic studies have also associated several polymorphism to a reduced response to ASA and clopidogrel [15]. Of interest, non-responders individuals have been associated with 5-fold greater risk of suffering CV events than those considered responders [39].
These two hypothesis previously exposed should be further approached in the clinical setting.
The ASA example
The efficacy of aspirin in preventing CVD appears to be lower in patients with diabetes mellitus (DM) compared to the general population, suggesting a lower clinical response to aspirin treatment in this population [40].
Henry, et al. have previously demonstrated a 24-hour time dependent aspirin loss of efficacy (ALE). Their study showed a 25% time-dependent increase in the so-called “non-responders” from the 2 hours to the 24-hours post ASA-administration. This ALE was observed mainly in patients with DM, high circulating levels of inflammatory markers, and active smokers [27]. Given the low rate of non-responders at 2-hours post-administration, it seems plausible to rule out any gene polymorphism involvement in the time-dependent increase.
We have previously reported significant variations in platelet response to ASA in a group of patients undergoing coronary artery bypass grafting (CABG). Prior the surgical procedure, a daily dose of ASA 100mg ASA was able to totally abolish Arachidonic Acid-induced WBI-aggregation. Surprisingly, after the procedure, all patients registered transient increases in platelet reactivity despite receiving the same ASA treatment. Up to 28% of post-CABG patients presented values of platelet aggregation similar to a group of non-treated healthy subjects that served as control group. These alterations that picked at seven days post-surgery were normalized one-month after CABG in all patients except for two that showed persistent signs of inflammation and thrombocytosis.
Reduced platelet inhibition to ASA was significantly associated with a daily platelet turnover higher than 10%. An association between increased IL6 levels and increased platelet count was also observed [23].
One of the strategies studied to improve the reduced antiplatelet effects of ASA was to increase the daily dose; but it has not been associated with a better prognosis [41,42].
It may be due to the fact that new platelets are released after the metabolization of ASA and they are not inhibited despite increasing the once-a-day dose of ASA.
Di Minno, et al. first suggested that the synthesis and release of new platelets into the circulation permits the recovery of thromboxane formation as early as 4 hours after aspirin ingestion [43].
The half-life of ASA in plasma is 15 to 20minutes, [44] thus, the time aspirin is present in the plasma to interact with circulating platelets is around 2 hours. In normal conditions, approximately 10% of circulating platelets are replaced daily, and near normal platelet aggregation to ASA is already found 48 to 72 hours after the last aspirin dosing [26]. The new platelets are able to synthesize thromboxane A2 (TXA2), and once the new platelets reaches 20% to 30% of all platelet count, it triggers aggregation of all the platelets including the older-acetylated ones, since these platelets remain sensitive to TXA2 even if they do not produce it [45]. This recovery could even be reached in less than 24 hours in certain pathological conditions characterized by an accelerated platelet turnover such as DM, smoking, advanced atherosclerotic lesions, and/or inflammatory syndrome [46].
There are two isoforms of enzyme cyclo-oxygenase (COX): COX1 and COX2. The antiplatelet effect of aspirin is mediated by the irreversible inactivation of COX-1, leading to the prevention of thromboxane A2 generation, a potent vasoconstrictor, platelet agonist and vascular smooth muscle cell mitogen [47,48]. However ASA is also a dose-dependent inhibitor of the prostacyclin production which fulfills physiological processes such as protection of the gastric mucosa, anti-aggregation and vasodilating effect at endothelial level [49].
COX-1 is rapidly resynthesized by nucleated cells; therefore the effect of ASA on nucleated cells (vg endothelial cells) lasts only for a relatively short time. In contrast, the effect of ASA on anucleate cells like platelets lasts for lifetime (7–10 days).
The COX-2 isoform is expressed in inflammatory states in response to oxygen reactive species, endotoxins, cytokines, or growth factors and can be found in newly formed platelets [50].
Aspirin is 170 times less potent as an inhibitor of the COX-2 enzyme compared to COX-1. In consequence, COX-2 requires higher doses of aspirin for inhibition and to consequently obtain anti-inflammatory effects, higher than 1.2 g. Therefore, low doses of ASA achieve adequate antiplatelet effect but a negligible anti-inflammatory effect. Using lower doses, it could prevent this imbalance and thus stimulate aspirin antiplatelet action and prevents gastrointestinal bleeding. In healthy people 95% suppression of platelet COX-1 activity is achieved with low-dose ASA (as low as 30 mg) [51,52].
From the currently available clinical data, the American College of cardiology/American Heart Association guidelines recommend 75 to 81 mg/d of ASA in the setting of CVD prevention because higher doses are not associated with increased event prevention, while increasing the bleeding risk. The recommendation in secondary prevention is to use aspirin 75 to 100 mg when it is associated with another antiplatelet, especially when it is considered for longer than 1 year [53].
Focusing on dosing, Perneby, et al. suggested that aspirin once per day might be insufficient in patients with increased platelet turnover because of incomplete suppression of platelet function over 24 hours [25].
It is hypothesize, that more frequent administration of even lower doses of aspirin could decrease ALE [52]. Dillinger, et al. demonstrated that the same dose of aspirin given twice per day was more effective than when given once a day to diabetic patients with elevated inflammatory markers or smoking [54].
We have demonstrated in post-CABG patients that low doses of ASA (100 mg) every 8 hours was a better strategy to prevent variations in anti-aggregation levels than a single higher daily dose (300 mg daily) [55] (Figure 2).
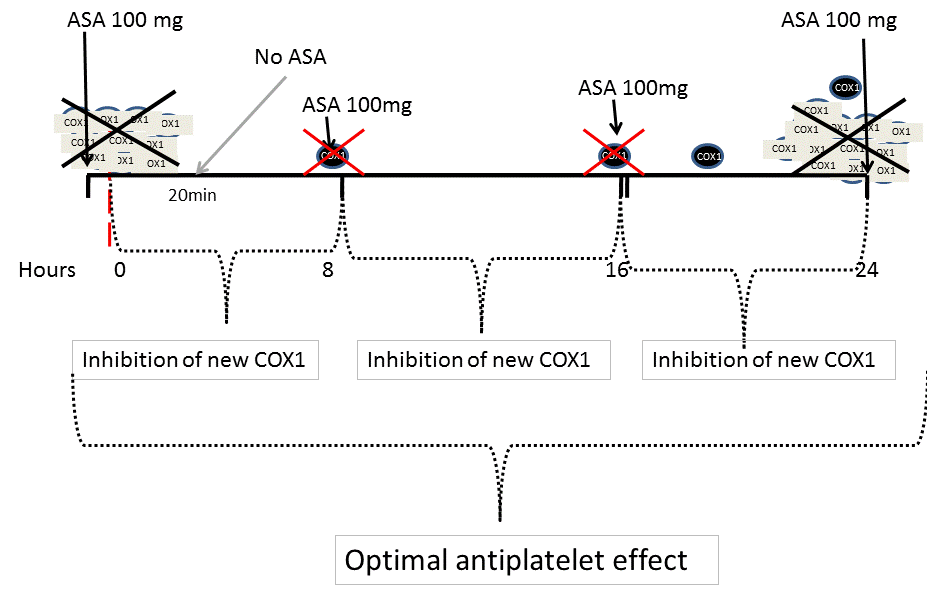
Figure 2. Low doses of ASA every 8 hours could be a better strategy than a single higher daily dose.
When double antiplatelet treatment became not enough
Following an ACS, there is a substantial risk for recurrent CV events despite ASA therapy. The CURE trial [11] showed that the addition of clopidogrel to ASA significantly reduces the risk of the composite outcome of death from cardiovascular causes, nonfatal myocardial infarction or stroke, during one year after an ACS with an increased risk of bleeding. Despite these benefits, many patients continued to have recurrent atherothrombotic events while receiving standard dual antiplatelet therapy.
More potent platelet inhibitor agents have showed a reduction in thrombotic events in subgroups known to have a more potent inflammatory state: diabetics and STEMI [12]. In fact, Prasugrel and Ticagrelor administration has significantly diminished the incidence of non-responders to their therapy [12]. This beneficial effect was accompanied by an increase in the rate of bleeding complications.
The same as ASA happens: after the first hours of contact with antiplatelet drugs, new young platelets have no P2Y12 blockade in the inter-dose phase. Whether the same situation applies to the newer and more potent antiplatelet agents is not well delineated yet (Figure 3).
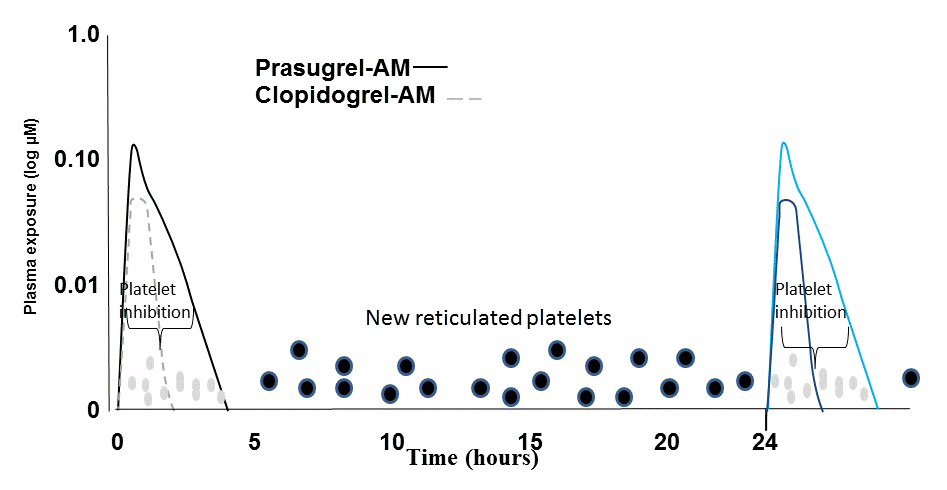
Figure 3. Once a day treatment. P2Y12 inhibitors lasts 2-4 hours. New platelets are “free” to aggregate in the inter-dose interval.
The optimal treatment of patients with NSTEACS must take into account the benefits of a more intense antiplatelet therapy with the increased risk of bleeding complications after the administration of higher doses. It is important to remember the strong association of bleeding with both, early and late mortality in NSTE-ACS patients [56].
Glycoprotein IIb/IIIa receptor inhibitors were tested trying to block another mechanism of activation and aggregation of platelets (Figure 4).
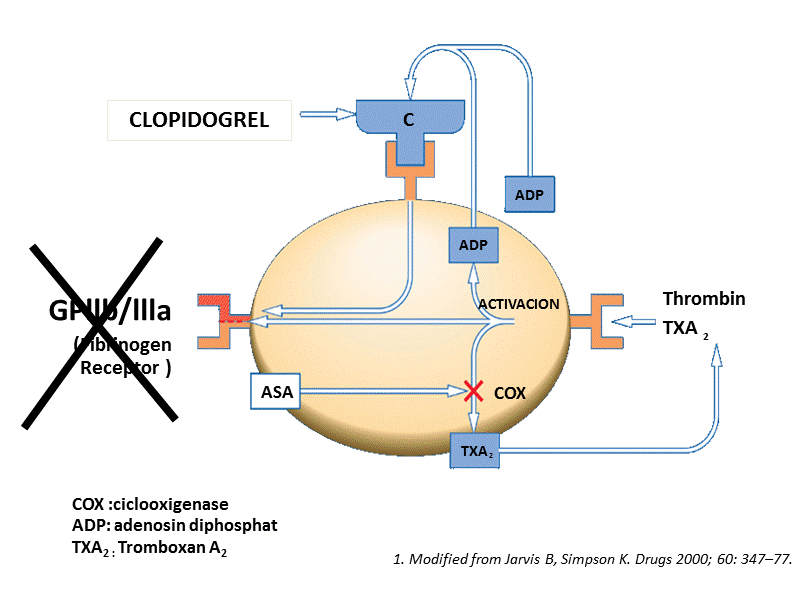
Figure 4. Blockade of platelet aggregation via GpIIbIIIa inhibition.
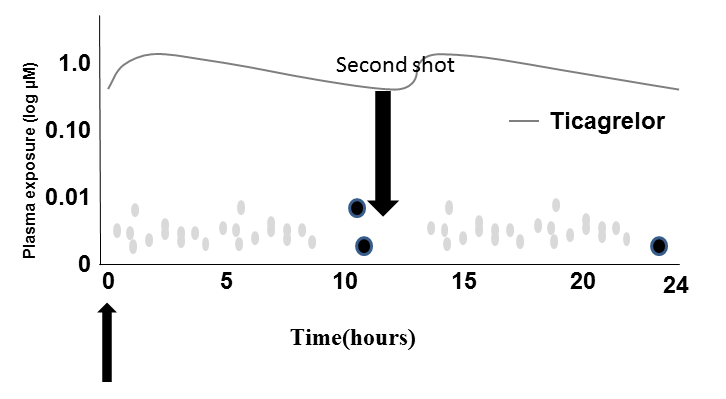
Figure 5. Antiplatelet plasma activity of Ticagrelor.
Figure 6. Pharmacodynamics of Ticagrelor.
In NSTE-ACS early glycoprotein IIb/IIIa inhibitors therapy are associated with significant reduction in ischemic events, largely driven by recurrence of ischemia or unplanned repeat revascularization, compared to a deferred therapy after coronary angiography, a benefit which is largely offset by an increased risk of major bleeding with this approach. As such, there is no difference in mortality between these two strategies.
The routine use of upstream GP IIb/IIIa inhibitors should therefore be reserved for patients at high ischemic and/or low hemorrhagic risk [57]. ESC guidelines recommend the addition of GP IIb/IIIa receptor inhibitor among patients who are already treated with DAPT for high-risk PCI (elevated troponin, visible thrombus) if the risk of bleeding is low (IB) [58]. In STEMI, AHA guidelines [59] consider that it is reasonable (indication class IIa) to begin treatment with an intravenous GP IIb/IIIa receptor antagonist such as abciximab (Level of Evidence: A), high-bolus-dose tirofiban2 (Level of Evidence: B), or double-bolus eptifibatide 270 (Level of Evidence: B) at the time of primary PCI (with or without stenting or clopidogrel pretreatment) in selected patients with STEMI who are receiving unfractionated heparin (UFH).The adjunctive use of GP IIb/IIIa agents at the time of PCI is only recommended in an individual basis for large thrombus burden or inadequate P2Y12 receptor antagonist loading.
There appears to be a relation between subgroups with higher inflammatory process associated to higher platelet turn-over and the patients that have shown benefit with long term infusion of GPIIbIIa (diabetics, multivessel disease and STMI). It could be hypothesized that when the GPIIbIIIa with long half-life are infused during hours, they are more helpful than when we use potent antiplatelet drugs.
Is Ticagrelor really different?
Ticagrelor, a reversible and direct-acting oral antagonist of the P2Y12 platelet receptor, as compared with clopidogrel, significantly reduced the rate of death from vascular causes, myocardial infarction, or stroke, as compared to clopidogrel. These benefits were attain without an increase in major bleeding but with an increase in the rate of non–procedure-related bleeding [13].
Different exclusive mechanisms have been proposed to explain the prevention of cardiovascular mortality observed with ticagrelor:
Pharmacokinetic
Unlike clopidogrel and prasugrel, ticagrelor does not require metabolic activation for antiplatelet activity. This allows ticagrelor to achieve near maximal platelet inhibition (~80%) within 1 hour post-administration, and to achieve maximal IPA at 2 hours compared while clopidogrel requires of 7-8 hours for achieving maximal IPA.
On the other hand, consistent with its noncompetitive and reversible pharmacology, the rate of offset with ticagrelor is also significantly faster compared with clopidogrel.
Pharmacodynamics
Ticagrelor inhibits the cellular uptake of adenosine, likely by inhibition of ENT1 [10]. Because one of the biological effects of adenosine is to inhibit the function of platelets [12-14], which express A2A receptors (and, to a lesser extent, A2B), ticagrelor inhibits platelet function not only by antagonizing P2Y12 but also by increasing the levels of adenosine [60].
Ticagrelor does not increase the production of adenosine, it inhibits adenosine uptake, and it is interesting to notice that adenosine increases in ischemic areas [61]. Adenosine has shown several effects that may be beneficial for patients with ACS, including cardioprotection, vasodilatation, inflammatory regulation, and platelet function inhibition [62].
But it is also possible that twice a day dosing may also play a role the benefit observed with Ticagrelor. In subgroups where platelets persist activated for long term, longer duration of more-intensive antiplatelet therapy studies have suggested a benefit to a longer duration of more-intensive antiplatelet therapy [63], including a higher net benefit with lower twice daily doses [64].
Conclusions
In the last years, we have payed attention to the balance between the thrombotic versus hemorrhagic risk. We know that there are conditions in which the patients have inappropriate antiplatelet effect and it has been related to different genetic, pharmacodynamics and pharmacokinetic mechanisms. Different strategies have been tried to supply these phenomena: increasing doses, adding antiplatelet drugs with different mechanisms of action or changing to new more potent antiplatelet drugs.
However, the thrombotic cardiovascular events persisted elevated and we began to see more threatened bleeding. Perhaps the strategy should have been focused on what happened to platelets in inflammatory states, considering that it has been demonstrated that new reticulated platelets appear in blood in a higher than normal daily turn-over and they are potentially more “resistant” to antiplatelet drugs: this could be the key to try fragmented low doses to avoid the bleeding risk and to obtain more stable antiplatelet effect in the 24 hours dose interval.
Expert opinion
Despite intensive antiplatelet treatment, some patients develop recurrent coronary and vascular events. We believe that a relationship between increased platelet turn-over and a hyper-inflammatory status can explain these results. Inflammatory disorders are commonly associated with thrombocytosis and the release of an increased number of young platelets may not be effectively inhibited by the once-a-day administration of antiplatelet drugs.
Inflamed/activated endothelium interacts with platelets by the expression of adhesive proteins on the endothelium. Activated macrophages, T cells and mast cells, release several types of molecules facilitating plaque rupture and subsequent acute thrombus formation, leading to platelet aggregation and secretion. These events perpetuate the inflammatory interactions between platelets, endothelial cells, and leukocytes
The synthesis and release of new platelets into the circulation promotes the recovery of the thrombosis capacity. Aggregation ability recovery depends on the half-life of the antiplatelet drug in plasma. On the other hand, immature platelets contain measurable amounts of cytosolic messenger ribonucleic acid (mRNA) that is translationally active and participates most actively in the thrombotic process, attenuating the inhibitory effects of these agents.
We have previously reported significant variations in platelet response to aspirin (ASA) in a group of patients undergoing coronary artery bypass grafting (CABG). Reduced platelet inhibition to ASA was significantly associated with a daily platelet turnover higher than 10% and we found an association between increased IL6 levels and increased platelet count.
It has been demonstrated that the same dose of aspirin given twice per day was more effective than when given once a day to diabetic patients with elevated inflammatory markers. We observed that low doses of ASA (100 mg) every 8 hours was a better strategy to prevent variations in anti-aggregation levels than a single higher daily dose (300 mg daily) after CABG.
The same as ASA happens with prasugrel and ticagrelor. After the first hours of contact with antiplatelet drugs, new young platelets have no P2Y12 blockade in the inter-dose phase. In fact, in subgroups where platelets persist activated for long term, longer duration of more-intensive antiplatelet therapy studies have suggested a benefit to a longer duration of more-intensive antiplatelet therapy, including a higher net benefit with lower twice daily doses. It is also important to evaluate the balance between the benefit of the antithrombotic treatment and the bleeding risk.
We consider that the key could be the treatment with fragmented low doses of antiplatelet drugs to avoid the bleeding risk and to obtain more stable antiplatelet effect in the 24 hours dose interval. Current trials are evaluating this hypothesis and we are actively working on it.
Highlights box
- Recurrent cardiovascular events are frequent despite intense antiplatelet treatment.
- Inflammatory disorders are commonly associated with thrombocytosis.
- The synthesis and r2021 Copyright OAT. All rights reserv the circulation are associated with the inter-dose recovery of platelet aggregation capacity.
- Fragmented low doses of antiplatelet drugs can obtain more stable antiplatelet effect in the 24 hours dose interval avoiding the bleeding risk of higher doses or more potent treatments.
References
- Davi G, Patrono C (2007) Platelet activation and atherothrombosis. N Engl J Med 357: 2482-2494. [Crossref]
- Libby P (2001) Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 104: 365-372. [Crossref]
- Yayan J (2013) Emerging families of biomarkers for coronary artery disease: inflammatory mediators. Vasc Health Risk Manag 9: 435-456. [Crossref]
- Madjid M, Casscells SW, Willerson JT (2007) Biomarkers of inflammation as surrogate markers in detection of vulnerable plaques and vulnerable patients In: Wellens HJJ, Holmes DR Jr, editors. Cardiovascular medicine. 3rd ed. London: Springer-Verlag London Limited; p. 641-51.
- Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, et al. (1994) The prognostic value of C-reactive protein and serum amyloid A protein in severe unstable angina. N Engl J Med 331: 417-424. [Crossref]
- Sabatine MS, Morrow DA, Cannon CP, Murphy SA, Demopoulos LA, et al. (2002) Relationship between baseline white blood cell count and degree of coronary artery disease and mortality in patients with acute coronary syndromes:a TACTICS-TIMI 18 (Treat Angina dThrombolysis in Myocardial Infarction 18 trial) substudy. J Am Coll Cardiol 40: 1761-1768. [Crossref]
- Mueller C, Neumann FJ, Perruchoud AP, Buettner HJ (2003) White blood cell count and long term mortality after non-ST elevation acute coronary syndrome treated with very early revascularisation. Heart 89: 389-392. [Crossref]
- Blum A (2005) White blood cell count and the coronary anatomy in acute coronary events. Am J Cardiol 95: 159-160. [Crossref]
- Lindsberg PJ, Grau AJ (2003) Inflammation and infections as risk factors for ischemic stroke. Stroke 34: 2518-2532. [Crossref]
- Armstrong EJ, Morrow DA, Sabatine MS (2006) Inflammatory biomarkers in acute coronary syndromes: part I: introduction and cytokines. Circulation 113: e72-75. [Crossref]
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK (2001) The Clopidogrel in unstable angina to prevent recurrent events trial investigators. Effects of Clopidogrel in addition to Aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 345: 494-502. [Crossref]
- Wiviott SD, Braunwald E, McCabe C H, Montalescot G. Ruzyllo W, et al. (2007) Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 357: 2001-2015. [Crossref]
- Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, et al. (2009) Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361: 1045-1057. [Crossref]
- Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, et al. (2009) Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 360: 363-375. [Crossref]
- Dahl ML, Gunes A (2010) Implications of inter-individual differences in clopidogrel metabolism, with focus on pharmacogenetics. Pharmaceuticals (Basel) 3: 782-794. [Crossref]
- Feero WG, Guttmacher AE (2011) Genomics and drug response. N Engl J Med 364: 1144-1153. [Crossref]
- Yang Y, Lewis JP, Hulot JS, Scott SA (2015) The pharmacogenetic control of antiplatelet response: Candidate genes and CYP2C19. Expert Opin Drug Metab Toxicol 11: 1599-1617. [Crossref]
- Sarma J, Laan CA, Alam S, Jha A, Fox KAA, Dransfield I (2002) Increased platelet binding to circulating monocytes in acute coronary syndromes. Circulation 105: 2166-217. [Crossref]
- Kuliczkowski W, Greif M, Gasior M, Kaczmarski J, Pres D, Polonski L (2011) Effects of platelet and inflammatory system activation on outcomes in diabetic patients with ST segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Kardiol Pol 6: 531-537. [Crossref]
- Patel PB, Pfau SE, Cleman MW, Brennan JJ, Howes C, Remetz M, et al. (2004) Comparison of coronary artery specific leukocyte–platelet conjugate formation in unstable versus stable angina pectoris. Am J Cardiol 93: 410-413. [Crossref]
- Wu Y, Wu H, Mueller C, Gibson CM, Murphy S, et al. (2012) Baseline platelet count and clinical outcome in acute coronary syndrome. Circ J 76: 704-711. [Crossref]
- Arazi HC, Badimon JJ (2012) Anti-inflammatory effects of anti-platelet treatment in atherosclerosis. Curr Pharm Des 18: 4311-4325. [Crossref]
- Cohen Arazi H, Doiny D, SpampinatoTorcivia R, Grancelli H, Waldman SV, et al. (2010) Impaired anti-platelet effect of aspirin, inflammation and platelet turn-over in cardiac surgery. Interac Cardiovasc Thorac Surg 10: 863-867. [Crossref]
- Kaser A, Brandacher G, Steur W, Kaser S, Offner FA, Zoller H, et al. (2001) Interleukin 6 stimulates thrombopoiesis through thrombopoietin: role of inflammatory thrombocytosis. Blood 98: 2720-2725. [Crossref]
- Perneby C, Wallén NH, Rooney C, Fitzgerald D, Hjemdahl P (2006) Dose- and time-dependent antiplatelet effects of aspirin. Thromb Haemost 95: 652-658. [Crossref]
- Awtry EH, Loscalzo J (2000) Aspirin. Circulation 101: 1206-1218. [Crossref]
- Henry P, Vermillet A, Boval B, Guyetand C, Petroni T, et al. (2011) 24-hour time-dependent aspirin efficacy in patients with stable coronary artery disease. Thrombhaemost 105: 336-344. [Crossref]
- Cesari F, Marcucci R, Caporale R, Paniccia R (2008) Relationship between high platelet turnover and platelet function in high-risk patients with coronary artery disease on dual antiplatelet therapy. Thromb Haemost 99: 930-935. [Crossref]
- Hsu HC, Tsai WH, Jiang ML, Ho CH, Hsu ML, et al. (1999) Circulating levels of thrombopoietic and inflammatory cytokines inpatients with clonal and reactive thrombocytosis. J Lab Clin Med 134: 392-397. [Crossref]
- Senaran H, Ileri M, AltinbaÅŸ A, KoÅŸar A, Yetkin E, et al. (2001) Thrombopoietin and mean platelet volume in coronary artery disease. Clin Cardiol 24: 405-408. [Crossref]
- Ly HQ, Kirtane AJ, Murphy SA, Buros J, Cannon CP, et al. (2006) Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 98: 1-5. [Crossref]
- Katoh A, Ikeda H (2012) Platelet count as a prognostic marker in patientswith acute coronary syndromes. Circ J 76: 591-592. [Crossref]
- Mangalpally KK, Siqueiros-Garcia A, Vaduganathan M, Dong JF, Kleiman NS, Guthikonda S (2010) Platelet activation patterns in platelet size sub-populations: differential responses to aspirin in vitro. J Thromb Thrombolysis 30: 251-262. [Crossref]
- Fager AM, Wood JP, Bouchard BA, Feng P, Tracy PB (2010) Properties of procoagulant platelets: defining and characterizing the subpopulation binding a functional prothrombinase. Arterioscler Thromb Vasc Biol 30: 2400-2407. [Crossref]
- Booyse F, Rafelson ME Jr (1967) In vitro incorporation of amino-acids into the contractile protein of human blood platelets. Nature 215: 283-284. [Crossref]
- Guthikonda S, Alviar CL, Vaduganathan M, Arikan M, Tellez A, et al. (2008) Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel inpatients with stable coronary artery disease. J Am Coll Cardiol 52: 743-749. [Crossref]
- Rowley JW, Schwertz H, Weyrich AS (2012) Platelet mRNA: the meaning behind the message. Curr Opin Hematol 19: 385-391. [Crossref]
- Ibrahim H, Schutt RC, Hannawi B, DeLao T, Barker CM, Kleiman NS (2014) Association of immature platelets with adverse cardiovascular outcomes. J Am Coll Cardiol 64: 2122-2129. [Crossref]
- Price MJ, Endemann S, Gollapudi RR, Valencia R, Stinis CT, Levisay JP, et al. (2008) Prognostic significance of post-clopidogrel platelet reactivity assessed by a point-of-care assay on thrombotic events after drug-eluting stent implantation. Eur Heart J 29: 992-1000. [Crossref]
- Belch J, MacCuish A, Campbell I, Cobbe S, Taylor R, Prescott R, et al. (2008) The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 337: a1840.
- Mehta SR, Tanguay JF, Eikelboom JW, Jolly SS, Joyner CD, et al. (2010) Double-dose versus standard-dose clopidogrel and high-dose versus low-dose aspirin in individuals undergoing percutaneous coronary intervention for acute coronary syndromes (CURRENT-OASIS 7): a randomised factorial trial. Lancet 376: 1233-1243. [Crossref]
- Antithrombotic Trialists' Collaboration (2002) Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 324: 71-86.
- Di Minno G, Silver MJ, Murphy S (1983) Monitoring the entry of new platelets into the circulation after ingestion of aspirin. Blood 61: 1081-1085. [Crossref]
- Patrono C, García Rodríguez LA, Landolfi R, Baigent C (2005) Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 353: 2373-2383. [Crossref]
- Patrono C, Rocca B (2007) Drug insight: aspirin resistance--fact or fashion? Nat Clin Pract Cardiovasc Med 4: 42-50. [Crossref]
- Brown AS, Hong Y, de Belder A, Beacon H, Beeso J, et al. (1997) Megakaryocyte ploidy and platelet changes in human diabetes and atherosclerosis. Arterioscler Thromb Vasc Biol 17: 802-807. [Crossref]
- Patrono C (1994) Aspirin as an antiplatelet drug. N Engl J Med 330: 1287-1294. [Crossref]
- Undas A, Brumel-Ziedins KE, Mann KG (2007) Antithrombotic properties of aspirin and resistance to aspirin: beyong strictly antiplatelet actions. Blood 109: 2285-2292. [Crossref]
- Fuster V, Sweeny JM (2011) Aspirin: a historical and contemporary therapeutic overview. Circulation 123: 768-778. [Crossref]
- Weber AA, Zimmermann KC, Meyer-Kirchrath J, Schrör K (1999) Cyclooxygenase-2 in human platelets as a possible factor in aspirin resistance. Lancet 353: 900. [Crossref]
- Reilly IA, FitzGerald GA (1987) Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood 69: 180-186. [Crossref]
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR (2007) Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 297: 2018-2024. [Crossref]
- Levine GN, Bates ER, Bittl JA, Brindis RG, Fihn SD, Fleisher LA, et al. (2016) 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease. A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation 133: e123-155. [Crossref]
- Dillinger JG, Drissa A, Sideris G (2012) Biological efficacy of twice daily aspirin in type 2 diabetic patients with coronary artery disease. Am Heart J 164: 600-606. [Crossref]
- Cohen Arazi H, Carnevalini M, Falconi E (2012) The association of antiplatelet aggregation effect of aspirin and platelet count. possible dosage implications. Rev Argent Cardiol 80: 114-120.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, et al. (2006) Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 114: 774-782. [Crossref]
- Sciahbasi A, Biondi-Zoccai G, Romagnoli E, Valgimigli M, Rasoul A, et al. (2012) Routine upstream versus selective downstream administration of glycoprotein IIb/IIIa inhibitors in patients with non-ST-elevation acute coronary syndromes: A meta-analysis of randomized trials. Int J Cardiol 155 (2012) 243–248. [Crossref]
- Hamm CW, Bassand JP, Agewall S, Bax J, Boersma E, et al. (2011) ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation The Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 32: 2999-3054. [Crossref]
- O'Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of st-elevation myocardial infarctiona report of the american college of cardiology foundation/ american heart association task force on practice guidelines. Circulation. 127: e362-e425. [Crossref]
- Nylander S, Femia EA, Scavone M, Berntsson P, Asztély AK, et al. (2013) Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. J Thromb Haemost 11: 1867-1876. [Crossref]
- Fredholm BB (2011) Physiological and pathophysiological roles of adenosine. Sleep Biol Rythms 9: 24-28.
- Armstrong D, Summers C, Ewart L, Nylander S, Sidaway JE, van Gieze JJJ (2014) Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J Cardiovasc Pharmacol Ther 19: 209. [Crossref]
- Yeh RW, Kereiakes DJ, Steg PG, Windecker S, Rinaldi MJ, Gershlick AH, et al. (2015) DAPT Study Investigators. Bene?ts and risks of extended duration dual antiplatelet therapy after PCI in patients with and without acute myocardial infarction. J Am Coll Cardiol 65: 2211-2221.
- Bonaca MP, Bhatt DL, Cohen M, Steg PG, Storey RF, et al. (2015) Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 372: 1791-1800. [Crossref]