Key words
SNP array, next generation sequencing, melanoma, biphasic cutaneous carcinomas, in situ hybridization
Abstract
The last decade has witnessed revolutionary developments in novel molecular techniques. The numerous available platforms have enhanced our diagnostic and prognostic capacities. Herein, we review the different applications of the currently available molecular tests in the field of neoplastic dermatopathology. First, we will discuss diagnostic platforms such as fluorescent in situ hybridization (FISH), comparative genomic hybridization (CGH), and RNA-based gene expression signature tests. We then consider the prognostic and theranostic value of platforms including high-throughput next-generation sequencing (NGS), matrix-assisted laser dissociation time of flight mass spectrometry (MALDI-TOF MS), and the gold standard of Sanger sequencing. Using melanoma, diagnostically challenging spitzoid lesions, and biphasic cutaneous carcinosarcomas as prototype neoplastic diseases, we will show the effects of these platforms on treatment development according to specific identified targets.
Introduction
Since the sequencing of nearly the entire human genome was first accomplished in 2003 the world of molecular pathology has experienced an exponential evolution [1]. This process evolved from karyotyping (in which whole chromosomes are discernible) to fluorescence in situ hybridization and comparative genomic hybridization (CGH, with which specific megabase regions are visualized), array-based CGH (aCGH), examining hundreds of base pairs and identifying single-nucleotide polymorphisms (SNP), and next-generation sequencing (providing single base pair resolution) [1]. Whole genome next-generation sequencing remains a cost-prohibitive method for many investigators, although considerable effort has been made to lower its cost for better incorporation into clinical practice. With the advent of newer technologies the cost of aCGH decreased recent years and the resolution of genome mapping increased [2]. A similar cost reduction has been seen in targeted next-generation sequencing, which has gained tremendous popularity and become the sine qua non for standard testing in personalized medicine centers.
Understanding the specific needs of the patient population in each practice and tailoring the different available platforms in that regard, remains the most important point that needs to be taken in consideration when implementing next-generation sequencing techniques. In this review, we will focus on clinical platforms that have shown promising results in diagnosis, prognosis, and theranostics in the clinical practice in dermatopathology. Data generated by these methods will enable pathologists and clinicians to better understand the numerous challenging cases and to design treatment plans customized to the patient’s specific tumor. We will not discuss available techniques that are currently limited to research use (whole-genome sequencing, non-targeted RNA sequencing).
From a practical standpoint, the different molecular platforms in clinical dermatopathology can be divided in 2 main categories: diagnostic and prognostic platforms and platforms for therapeutic guidance. The diagnostic and prognostic platforms include fluorescence in situ hybridization (FISH), comparative genomic hybridization and SNP array (CGH+SNP) array, and gene expression signature (quantitative reverse transcriptase PCR). Platforms for therapeutic guidance include Sanger sequencing, matrix-associated laser di-ionization time of flight mass spectrometry (MALDITOF-MS), and high-throughput targeted next-generation sequencing (NGS). Each of these platform categories is discussed below.
Fluorescence in situ hybridization
Conventional cytogenetics and CGH paved the way for FISH in dermatopathology. The initial data published by Bastian et al. in 2002 showed recurrent genomic aberrations in melanomas involving chromosomes 9, 10, 7, and 6, in contrast to most Spitz nevi, which do not show aberrations [3]. The following years witnessed a plethora of publications addressing the application of FISH and its diagnostic and prognostic value in melanocytic lesions.
FISH uses fluorescent-labeled probes targeting specific genomic areas of interest. Hybridization of these probes is performed on baked slides prepared from formalin-fixed, paraffin-embedded (FFPE) tissue at 37°C for 16 to 18 hours using an automated co-denaturation oven (HYBrite or ThermoBrite Denaturation/Hybridization System, Abbott Molecular) [4].
In 2009, Gerami et al. published their experience with the application of FISH in nevoid melanomas and nevi using a set of 4 probes targeting ras responsive element-binding protein-1 (Vysis LSI RREB1-Spectrum Red), myeloblastosis (Vysis LSI MYB-S Gold), cyclin D1 or chromosome 11q (Vysis LSI CCND1-Spectrum Green), and centromeric enumeration probe control for chromosome 6 (Vysis LSI CEP6-Spectrum Aqua) from Abbot Molecular [4]. In a series of 51 benign nevi and 51 nevoid melanomas, the authors showed that this set probe has a sensitivity of 86.7% and a specificity of 95.4%when used to discriminate between histologically unequivocal melanomas and benign nevi. The limitations of this study included a low sensitivity in differentiating between spitzoid melanomas and benign Spitz nevi (reported sensitivity of 70% in melanoma with spitzoid morphology), as well as a limited capacity to differentiate true gains from tetraploidy, which may cause difficulties in interpretation by inexperienced examiners. In addition, the cases included were unequivocal (i.e., they had enough histological criteria to be able to make a diagnosis of benign or malignant melanoma) [4]. It is important to take into consideration that essentially most applications of FISH will target equivocal rather than unequivocal lesions. In 2012, the same group published the results of a refined set of probes testing 322 tumors, including 152 melanomas and 170 nevi. In a cohort of 51 melanomas and 51 nevi the probe set targeting 9p21, 6p25, 11q13, and 8q24 probe set showed a sensitivity of 94% and specificity of 98%. This is significantly higher than previous sensitivity and specificity (75% and 96%, respectively) obtained in the same cohort with the initial probe set [5]. One of the main advantages of this refined probe set is that it targets 4 genomic loci on 4 different chromosomes, resulting in easier interpretation for examiners and a lower false positive rate due to tetra/polyploidy (Figure 1). Subsequent studies showed that gains in MYC at 8q24 are associated with an aggressive clinical course [6]. A multicentric case-controlled study involving multiple melanoma treatment centers in the United States and Australia showed that homozygous 9p21 deletion was highly associated with clinically aggressive behavior (p<0.0001) and death due to disease (p=0.003) in atypical spitzoid tumors [7]. On the basis of the results of these 2 studies and previous studies by Gerami et al. [5,7]. FISH panels became commercially available. One popular commercially available panel is NeoSITE Melanoma by Neogenomics. Using previously published cutoffs, this panel showed a sensitivity of 70% and specificity of 98% [8]. One of the major shortcomings in the validation studies of these probes was the case selection. In most of the initial studies, investigators used clear-cut malignant or benign tumors in their validation cohorts. In a study published in 2014, North et al. reviewed 804 cases of ambiguous melanocytic lesions tested using the first-generation FISH panel (targeting 3 loci on chromosome 6 and 1 on 11q) and showed that this panel assisted in reaching a final definitive diagnosis in 88% of the cases, with the remaining 12% having an ambiguous diagnosis even with FISH studies (equivocal FISH results) [9]. They showed a sensitivity of 78% and a specificity of 94%. A recent study in a major cancer center reviewed 37 cases of ambiguous melanocytic lesions using the second-generation NeoSITE FISH panel and found a sensitivity of 39% and specificity of 89% [10]. In conclusion, albeit fairly specific in ambiguous melanocytic lesions, FISH sensitivity varies depending on the morphology (spitzoid vs. nevoid vs. spindle). Additional limitations include the need for experienced technologists when interpreting the findings. Pathologists and clinicians should be aware of these limitations when incorporating testing results into their final assessment of diagnostically challenging melanocytic lesions.
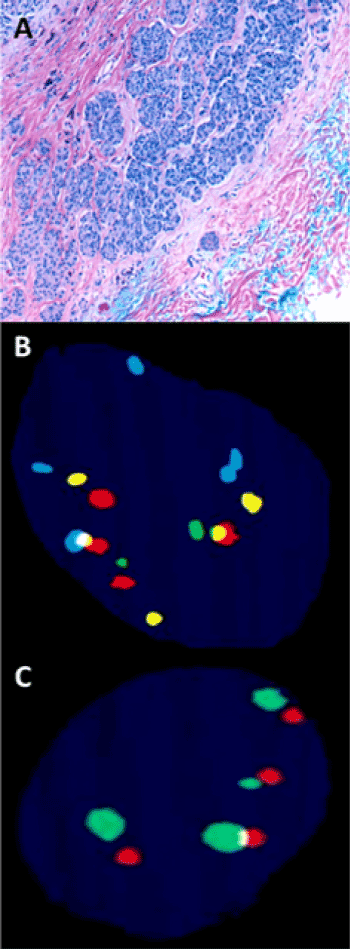
Figure 1. An example of a challenging spitzoid lesion on the right breast of a 22-year-old woman (A-C). A. Hematoxylin-eosin, 10× magnification. Notice the lack of maturation and expansile pattern of growth in the deeper aspects of the lesion (blue ink). There was moderate cytologic atypia but no mitotic activity. B, C. FISH studies were performed using probes targeting the following loci: 6p25(RREB1) spectrum blue, 6q23 (MYB) spectrum yellow, CEP 6 spectrum red, 11q13(CCND1) spectrum green, 9p21(CDKN2A) spectrum green in part C as well as CEP 9 (centromeric probe) spectrum red in part C. The final count based on 30 consecutive tumoral cells showed the following ratios: gain in 6p25 (RREB1) relative to CEP6 = 23% (cutoff >55%), gain in 6p25 (RREB1) = 60% (cutoff >29%), loss in 6q23 (MYB) relative to CEP6 = 27% (cutoff >40%), gain in 11q13 (CCND1) = 47% (cutoff >38%). Notice that 2 of the absolute numeric values of the count meet the criteria for a “positive result.” However, review of the actual slides is of the highest importance as it shows “homogenous” amplification across all probes targeting different loci. Furthermore, when looking at probes targeting 9p21 and CEP9 in part C, one can see that both have similar numbers of amplified signals with a ratio of 9p21/CEP = 1 and without evidence of homozygous or heterozygous loss in the 9p21 locus. These findings are consistent with a polyploidy pattern, a common source of false positive result for inexperienced reviewers. This case had a negative FISH result; the final rendered diagnosis, given the moderate cytologic atypia, was Spitz nevus with atypical features.
Genome-wide comparative genomic hybridization (CGH) with single-nucleotide polymorphism (SNP) array
This recent scalable and highly specific technique shares certain similarities with conventional CGH. This high-resolution platform is based on the molecular inversion probe technology and is suitable for highly degraded FFPE samples (short probe interrogation site of just 20-45 base pairs). Briefly, a short circular fluorescent-tagged probe targeting a specific genomic area anneals to the target sequence using target complementary region 1 (5′ end) and target complementary region 2 (3′ end). In the presence of ligase and polymerase, the next step is initiated with gap filling and polymerization in order to synthetize the complementary DNA fragment to the targeted sequence. The probe release cleavage site is then activated, which enables a linear/flat configuration of the probe/target tandem. This is followed by a target enrichment step using a simple PCR step with 2 primers present within the probe design (PCR primer 1: forward 5′ primer, and PCR primer 2: reverse 3′ primer). The enriched targets are then hybridized to a SNP chip (matrix containing complementary DNA sequences to the enriched target), which leads to a release of the fluorophore tag that enables signal detection [11-13]. The signal intensity is then converted to a log2 ratio scale for data interpretation. CGH+SNP array permits the use of highly fragmented DNA, with relatively low sample input (80 ng of FFPE-derived DNA). One of the most important advantages of CGH+SNP array is a very high-resolution whole-genome detection of copy number changes and copy neutral aberrations, such as loss of heterozygosity (LOH) and uniparental disomy (UPD), on the same array [13].
Multiple commercially available platforms, using the same principle described above, have been developed by 2 main companies ,Agilent and Affymetrix, and are available for researchers and clinical laboratories. The platform designs differ mainly in the targeted genes (cancer genes vs. constitutional genes). Each platform provides numerous choices of SNP chips [12,13]. The main difference between these chips is the density of the oligoprobes used: a higher probe density provides a higher resolution for both tested SNPs and copy number variants in cancer genes. This is also reflected in the spacing between the probes: the less spaced the probes are, the denser the coverage and the higher the resolution. In 2003, Bastian et al. used conventional CGH to show a pattern of chromosomal aberration in melanomas that is distinct from that of melanocytic nevi [14]. Melanomas showed recurrent genomic aberrations involving chromosomes 5p, 11q, 12q, 13q, 15, and 17p. None of the tested nevi showed genomic aberrations except for desmoplastic Spitz nevi, which showed constantly focal 11p gains [14]. Another more recent series by Chandler et al. investigated the sensitivity and specificity of CGH+SNP array in challenging melanocytic lesions. These authors found that Affymetrix’s Oncoscan FFPE platform has an 89% sensitivity and 100% specificity in non-ambiguous lesions. Nevertheless, the sensitivity and specificity of the assay were very limited in the 11 ambiguous lesions tested, with at least 3 cases showing discordance between the CGH+SNP call and the clinical follow-up [15]. On the basis of our experience, CGH+SNP analysis is useful mainly in melanocytic lesions with worrisome histological features where both immunohistochemical and FISH analysis fail to provide supportive findings to render a melanoma diagnosis. In these cases, CGH+SNP could show genomic aberrations in different loci that are not targeted by the available FISH panels. Another important limitation that should be taken into consideration with CGH+SNP array is its relatively high cost compared with other traditional targeted methods (i.e., FISH or conventional CGH).
Gene expression signature (quantitative reverse transcriptase (qRT)-PCR)
Gene expression signature using quantitative reverse transcriptase PCR was introduced in the armentarium of diagnostic methods for challenging melanocytic lesions by Clark et al. [16,17]. In a first discovery phase, the authors tested 31 melanomas and 56nevi. On the basis of an extensive literature search and whole-genome sequencing data, the authors chose 79 genes that showed significant differential expression in melanomas compared with normal tissue [16]. The 79 genes were tested by qRT-PCR in their discovery cohort. After RNA extraction from FFPE tissue, RNA was transformed into cDNA using reverse transcriptase. Specific primers for each gene were then added and PCR cycling was initiated. The quantitative expression value for each gene was measured by determining the crossing threshold (CT) of each gene. All experiments were conducted in triplicate to confirm reproducibility [17]. The CT of each gene for each sample replicate was normalized by the average CT of housekeeper genes on the same replicate, yielding ΔC (CT of the gene of interest minus CT of the housekeeping gene). Using receiver operator curve analysis, the authors selected genes with an area under the curve of >70%. This analysis yielded a list of 40 genes, which were included in the second verification phase [17]. This phase included 454 melanocytic lesions, including 244 melanomas and 210 benign nevi. qRT-PCR was performed using specific primers for the 40 genes of interest with ΔC as a marker of expression for each gene. This was followed by forward selection of important genes using a logistic regression model to identify the subset of genes that most effectively distinguishes benign from malignant lesions. This analysis identified a set of 14 genes that showed the most effective differentiating power in the forward selection: melanocytic differentiation PRAME (preferentially expressed antigen in melanoma), the S100A9-related genes (S100 A7, S100 A8, S100 A9, S100 A12, PI3), and the immune group genes (CCL5, CD38, CXCL10, CXCL9, IRF1, LCP2, PTPRC, SELL) (18). A refined logistic regression model was then used to generate a single melanoma diagnostic score (MDS) capable of differentiating benign nevi from malignant melanomas [17]. The generated scores ranged from -16.7 to -2.1 for the benign category, MDS from -2.0 to 0.1 defined the indeterminate category, and scores 0.2 to +11.1 were reported as malignant [17,18]. This verification phase was followed by the third phase, the validation study. This phase included 437 melanocytic lesions, including 211 melanomas and 226 nevi. Using the same methods and the established score for the 14 mentioned genes in addition to 9 housekeeping genes, the study showed a sensitivity of 90% and a specificity of 91% [18]. The clinical influence of this gene expression panel on diagnosis in daily dermatopathology practice is currently the subject of a multicenter prospective study including 96 dermatopathology practices. Preliminary analysis of 1695 submitted cases (including 218 ambiguous/challenging cases) showed a significant reduction from 80.3% to 37.6% in the number of cases that were initially recorded with a pre-test diagnosis of indeterminate [19]. In 39.4% of cases receiving a benign MDS result, recommendations were downgraded to less invasive treatment. In 45.8% of cases receiving a malignant MDS result, recommendations were upgraded to a more invasive treatment [19]. These findings suggest that this gene expression panel can be helpful when appropriately used in an adequate clinical setting. It should be mentioned that this testing is reserved for first-time biopsies. Patients with a history of immunosuppressant therapy, radiation treatment, re-excised specimens, and metastatic melanomas are not candidates for testing [19].
Sanger sequencing
In 1977, Fred Sanger and Alan R. Coulson published 2 papers on the rapid identification of DNA sequences [20,21], which served as the basis for deciphering genes and genomes and for current next-generation sequencing [22]. The simplicity of this method and the reduction of handling toxic chemicals made “Sanger sequencing” the most frequently applied DNA sequencing method for more than 25 years. Up to now, it remains the “gold standard” in routine diagnostics focused on specific analysis of one or a few genes. Limitations of this technique include its low sensitivity (analytical sensitivity of approximately 20%, i.e., it cannot detect mutations with tumor content less than approximately 40%), which limits its use in biopsies or cytological specimens given the potential risk of false negative.
At present, the National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology (ESMO) evidence-based guidelines are not recommended for mutational testing of primary tumors without metastasis [23,24]. In contrast, such testing is mandatory to detect targetable mutations in patients with advanced disease (unresectable or metastasized stage III or stage IV) and is highly recommended in high-risk resected disease (stage IIc, stage IIIb–IIIc). If the tumor shows a wild-type sequence for BRAF, subsequent testing for NRAS and KIT mutations should be considered [23,24]. Sequencing for these genes can be performed in a stepwise fashion by Sanger sequencing or in a one-step process using available extensive next-generation panels (see below).
Matrix associated laser di-ionization time of flight mass spectrometry
The Sequenom MassARRAY system, based on MALDI-TOF mass spectrometry, was originally designed to analyze SNPs in amplified DNA fragments but was then adapted in molecular oncology for detection of known well-defined mutations (single-nucleotide variants, SNV) [25,26]. The principle in this application is that mutant and germline alleles for a given point mutation produce single-allele base extension reaction products of different masses that are then resolved by MALDI-TOF Mass Spectrometry [26]. The assay consists of an initial locus-specific PCR reaction, followed by single base extension using mass-modified dideoxynucleotide terminators of an oligonucleotide primer which anneals immediately upstream of the polymorphic site of interest [26]. The Sequenom platform uses matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) to distinguish the products of primer extension reactions (performed on PCR products) in a sequence-specific manner based on the mass and charge [25,26]. Using MALDI-TOF mass spectrometry, the distinct mass of the extended primer identifies the SNP allele or the somatic single nucleotide variant [25,26]. In our experience, MALDITOF-MS is a very powerful platform; the automated workflow and relatively short hands enable multiplexing of multiple samples on one array chip with a total time of 9.5 hours from PCR setup to report generation. It has a lower limit of detection (analytical sensitivity is around 5%), a sensitivity that is significantly higher compared with conventional Sanger sequencing. Nevertheless, interpretation of results can be difficult at times, because salt complexes may cause background interference with the detected signals that could give false positive results (Figure 2). Overcoming this problem can be achieved by performing an exhaustive post-primer extension salt-cleaning step to ensure that no excess salt components are carried to the detector.
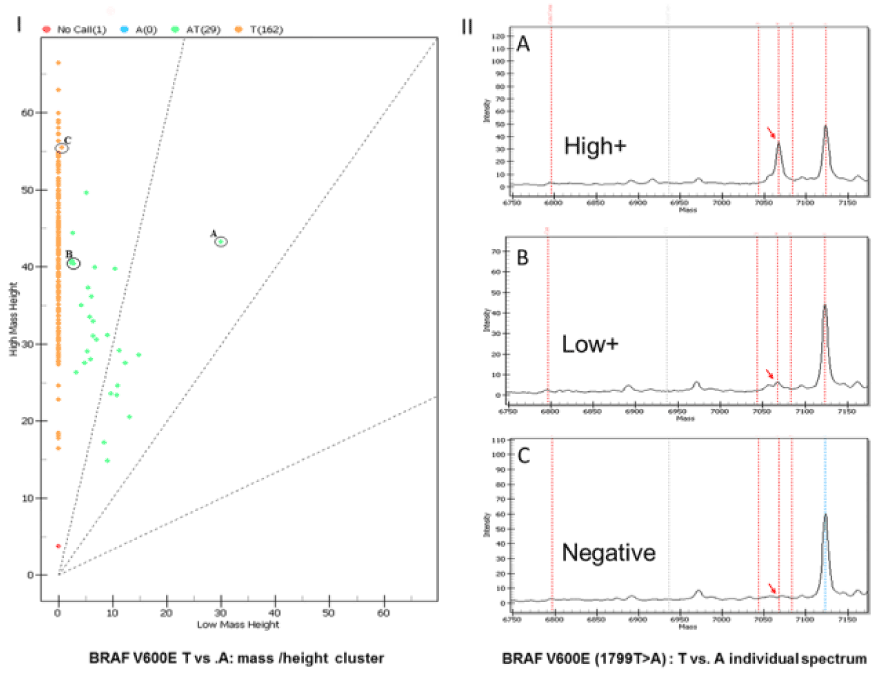
Figure 2. An example of 3 different melanoma cases with different tumor purity tested using the MALDITOF platform (specimens A, B, and C with decreasing tumor percentages of 60%, 20%, and 5-10%). I, The mass vs. height scatter plot showing the overall distribution of the 3 different samples based on the presence or absence of the detected single-nucleotide variant. Each orange point refers to a tested sample. Notice that negative sample fall altogether in a linear fashion with the same mass characteristic. Each orange dot indicates a case where the primer extension yielded a T base at position 1799 of the BRAF gene. This is the expected reference non-mutated base. All orange points refer to negative cases. Mutant samples in the run show a scatter depending on the tumor percentage. The green dots indicate different heterozygous mutation levels with substitution of the T at position 1799 with an A yielding a heterozygous genotype in the study sample (AT). Notice that the higher the tumor percentage is, the higher the change in the mass compared with the negative baseline is in the mutated sample (sample A vs. sample C). II, Individual spectra for the different melanoma samples with decreasing volume tumor. Notice that in sample A the wild-type extended base (T) has a mass different from that of the mutant base (A), enabling its detection. Also notice that the peak of the detected signal (intensity) is proportional to the tumor content, with the highest peak associated with the highest tumor volume. Note that in specimen C, a cautionary note should be added to the signed-out report mentioning that the tumor percentage was suboptimal and that a false negative result cannot be ruled out with certainty.
High-throughput targeted next-generation sequencing
High-throughput massive parallel sequencing, also known as next-generation sequencing (NGS) technology, can generate millions of reads in a relatively short time, making this a powerful tool for genome research [27-29]. For example, one full NGS run can produce 1 million to 5,000 million reads in 8 hours to 5 days, depending on the platform [29]. Such a high-throughput capability of genome sequencing or re-sequencing projects should efficiently and accurately discover and genotype many thousands of genetic polymorphisms, mainly SNPs, which can be used to investigate quantitative, functional, and evolutionary genomics in humans, animals, and plants.
There are 2 major commercially available methodological approaches in next-generation sequencing (Table 1) [30,31]. The amplicon approach is based on a series of multiplex PCR reactions leading to numerous fragments of DNA (amplicons) that are simultaneously sequenced on an ion semiconductor sequencer, enabling detection of numerous events including mutations (single nucleotide variants, in-frame deletions and insertions, and frame-shift events) as well as copy number variations (copy number losses and gains). The solid-phase hybrid-capture approach uses a very limited initial PCR amplification step in the library preparation, followed by hybridization to a solid phase with bridge amplification of the barcoded libraries. Sequencing by synthesis is performed using 4 different dyes with different dye terminators.
Table 1. Summary of different next-generation sequencing platforms available for clinical practices by different companies.
Manufacturer [30,31] |
Platform type |
Sequencing method |
Read
length |
Data range |
Time/run |
Accuracy* |
Illumina |
MiSeq |
Sequencing
by synthesis |
2 × 300 bp |
0,3-15 Gb |
5-65 hours |
>Q30 |
Illumina |
NextSeq 500 |
Sequencing
by synthesis |
2 × 150 bp |
30-120 Gb |
12-30 hours |
>Q30 |
Illumina |
HiSeq X |
Sequencing
by synthesis |
2 × 150 bp |
1800 Gb |
<3 days |
>Q30 |
Thermo
Fisher
Scientific |
Ion Torrent
Proton
System |
Ion
semiconductor |
2 × 200 bp |
Up to 10 Gb |
2-4 hours |
Q20 |
Thermo
Fisher
Scientific |
Ion Torrent
PGM 318 |
Ion
semiconductor |
400 bp |
1,2-2 Gb |
7,3 hours |
>Q20 |
Thermo
Fisher
Scientific |
SOLiD 5500
System |
Sequencing by ligation |
2 × 50 bp |
Up to 300 (80-100 GB average) |
12-16 days |
>Q30 |
This is not an exhaustive summary. For more details please visit the reference websites.
*Please refer to definitions below for a definition of Accuracy
Helpful definitions
Sequencing methods: Notice the 2 major sequencing methods mentioned in the text: sequencing by synthesis with dye termination (Illumina platforms) and sequencing by synthesis with ion semiconductor, which is based on the detection of hydrogen ions released during the polymerization of DNA (a.k.a. synthesis) (Thermofisher platforms). The released hydrogen leads to a change in the pH that is detected by an ion sensor, transforming it into an electrical signal that is captured. One of the limitations of the ion is homologous sequence homopolymer (i.e., GGGGGGGGG). These types of sequences lead to slippage and sequencing errors because such areas of the genome with homopolymeric sequences tend to have lower accuracy when sequenced using ion semiconductor methods. The new SOLiD platform by Thermofisher uses a universal primer and a set of fluorescent-labeled probes that are ligated to the target sequence (similar to the solid-phase method by Illumina) associated with periodic primer resetting, enabling repetitive sequencing of the same base and yielding a very high accuracy.
Accuracy: Accuracy in next-generation sequencing is measured using the Q score, also referred to as the Phred Quality score measure The higher the Q score, the higher is the base calling accuracy, i.e., the probability that the variant called on the sequencing is truly present and not a mere artifact. A Q score >30 is ideal, with a Q score >20 being acceptable (accuracies of 99.99% and 99%, respectively). Q scores are defined as a property that is logarithmically related to the base calling error probabilities (P): Q = −10 log10P. [
Read Length: The read length refers to the length of the sequence generated after the library preparation. A library consists of the generated hybrids of target fragmented sequences (usually down to 150 bp) with the complementary barcoded sequences provided by the manufacturer (commonly referred to as “baits”). Once the library is ready, pair end sequencing (meaning sequencing from 5′ to 3′ and 3′ to 5′) occurs, simultaneously generating a bi-directional read for each sequence. Notice that the more fragmented the DNA is, the harder it is to sequence it, as the small fragments yield suboptimal reads for realignments. DNA fragmentation happens frequently with time. This is one of the reasons why relatively new or recent specimens perform better in NGS. This read length also varies with the different types of platforms.
Data Range: All sequencers produce FASTQ files. FASTQ format is a text-based format for storing both the biological sequence (usually nucleotide sequence) and its corresponding quality scores (Q scores). The size of these files containing sequencing data that is converted into multiple of the unit byte for digital information (megabytes, gigabytes, and even terabytes). The MiSeq platforms have relatively limited capacity in the depth of sequencing. Hence they generate less data per run compared with higher performance machines such as HiSeq. This is important to consider depending on the application and the gene panel tested. In common clinical practice when performing NGS for targeted therapies using a targeted exon panel, MiSeq platforms are efficient and provide a rapid turnover. In research-oriented facilities that perform deeper sequencing across a more expansive set of genes (400-500 or even whole-genome sequencing), HiSeq sequencing systems provide a higher quality and quantity of data.
The limitations of NGS include the prohibitive cost when the projected testing volume does not justify its implementation. The other limitation is the amount of raw computational data requiring robust bioinformatics pipelines for analyses to extract specific information. Consequently, bioinformatics knowledge plays a key role in the investigation of data and detection of possible errors and reading artifacts [32]. Despite the high cost, NGS remains cost effective when used to analyze a high number of genes, and it is definitely more sensitive than conventional Sanger sequencing.
The advent of large-scale genome analysis through these NGS techniques led to The Cancer Genome Atlas (TCGA) project. This comprehensive joint effort of the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) aims to explore the molecular basis of cancer through the application of genomic analysis technologies [33]. In 2012, Hodis et al. published their experience with 130 melanomas including 95 melanomas of cutaneous origin, 5 of acral, 2 of mucosal, 1 of uveal, and 18 of unknown primary origin [34]. Using a computational permutation modeling analysis system, they identified numerous driver mutations in candidate genes in melanomas. A driver mutation is a genomic event that confers “fitness” to the cancer cell, promoting its survival and its escape from the host immune system. A passenger mutation, on the other hand, lacks these properties and would be present as a standard phenomenon (similar to what is seen in UV signatures and the plethora of single-nucleotide mutations) [35]. The genes identified in this study included the known mutations that lead to the constitutive activation of the mitogen-activated kinase (MAPK) pathway through mutation of BRAF or NRAS in addition to PTEN and TP53. Indeed, mutations in the serine-threonine kinase BRAF, particularly at codon 600 with substitution of valine by either glutamate (V600E) or lysine (V600K) or arginine (V600R), are present in about 50% of patients with cutaneous melanoma and in 10% to 20% of melanomas arising in mucosal or acral locations, but are entirely absent in uveal melanomas [36].
In addition to these results, 6 novel melanoma genes with driver mutations (PPP6C, RAC1, SNX31, TACC1, STK19, and ARID2) were identified. This study served as the basis for the TCGA melanoma database. In addition, it was the first scientific study that showed unequivocal evidence for a direct mutagenic role of UV light (mostly UVA) in melanoma pathogenesis [34].
A subsequent study that contributed to the TCGA database showed similar results in a cohort of 303 metastatic melanomas [37]. Both of these studies highlighted recurrent mutations in the RAC1, PPP6C and STK19 genes that were potentially targetable [34,37]. RAC1 (ras-related C3 botulinum toxin substrate 1) functions as a molecular switch that activates downstream proteins including PAK1 (p21-activated protein kinase 1). This promotes cellular adhesion, migration, and invasion [38]. RAC1 mutations occur in 3-5% of melanomas. These mutations show a hot spot pattern centered in the same codon, with most of them affecting the same nucleotide. The most frequent mutation is c.85C > T transition, resulting in a P29S amino acid change [34,37]. The change in the amino acid leads to a conformational change of the protein favoring the GTP-bound state (active state) over the GDP-bound state (inactive state). This leads to uncontrolled downstream signaling that leads to cell proliferation [34,37]. In vitro studies using melanoma cell lines showed that RAC1 P29S mutant cells are resistant to BRAF inhibitors (vemurafenib and dabrafenib) and have at least a partial sensitivity to MEK inhibitors (trametinib). These findings highlight the future potential benefit of MEK inhibitors in patients with wild-type BRAF melanomas with RAC1 P29S mutations [39].
A recent study using targeted next-generation sequencing in 140 spitzoid neoplasms (including spitzoid melanomas, atypical spitzoid tumors and Spitz nevi) showed recurrent Kinase fusions with potential targets for kinase inhibitors [40]. The most frequent gene rearrangements included ROS1 (17%), NTRK1 (16%), ALK (10%), BRAF (5%), and RET (3%), all of which resulted in in-frame kinase fusions. Using xenograft models, the authors showed that these kinase fusions are all oncogenic by inducing melanocytic tumors in mice [40]. These structural events were similar to fusions in lung adenocarcinomas [41]. Functional in vitro studies performed by the same group showed that cell lines with ROS1, ALK, NTRK1, and RET fusions showed increased phosphorylation of AKT and ERK, demonstrating activation of the MAPK pathway and at least partial inhibition of MAPK pathway signaling with different tyrosine kinase (TK) inhibitors [40]. These findings suggest that TK inhibitors that are FDA approved in fusion-positive lung adenocarcinoma (cabozantinib, crizotinib) can be considered for off-label use in patients with advanced and metastasizing spitzoid neoplasms that fail conventional treatment paradigms. Another important genomic rearrangement identified in melanoma with the NGS technique are BRAF translocations. BRAF translocations occur in pan-negative melanomas (4-5% of all melanomas) [42]. BRAF translocations constantly involve the last set of exons (exons 9-18), which contains the serine/threonine kinase activity. This leads to loss of a RAS-binding domain (RBD), leading to loss of the negative feedback loop on the serine threonine kinase domain [42]. Preclinical data using cell lines with PAPSS1-BRAF showed increased sensitivity to the MEK inhibitor trametinib but not to the BRAF inhibitor vemurafenib in MAPK pathway signaling [42]. These findings suggest that some melanomas with BRAF translocation may be sensitive to MEK inhibitors. While BRAF V600E remains the most common type of BRAF mutation seen in melanomas, other variants exist. Most of the targeted therapies that have been developed and validated in clinical trials have focused on this specific variant as well as BRAFV600K. Indeed, recent years have witnessed numerous FDA approvals of different targeted therapies that showed higher efficacy than conventional chemotherapy (dacarbazine) in advanced-stage and metastatic BRAF V600 mutant melanomas [43]. Relapse and resistance occur constantly in BRAF V600E melanoma patients with metastatic disease when treated with BRAF inhibitors [44]. The response rates vary from 40% in patients treated with vemurafenib as monotherapy to 59% in patients treated with dabrafenib as monotherapy [44]. [However, responses are generally short-lived, and resistance usually occurs within 8 months for vemurafenib [45,46] and within 8.8 months for dabrafenib [47]. Recent trials have focused on overcoming these resistance mechanisms by combining BRAF inhibitor therapy with pan-RAF or MEK inhibition (trametinib and cobimetinib) [48,49]. In the CombiD study, the progression-free survival was 8.8 months for patients treated with dabrafenib alone compared with 9.3 months for those treated with the combination of dabrafenib and trametinib [48]. Similar results are available also for the CoBRIM study, in which the combination of vemurafenib and cobimetinib improved progression-free survival to 9.9 months compared with 6.2 months for vemurafenib alone, at the cost of some increased toxicity [49]. This result has led to the recent approval by the FDA of combinational BRAF (dabrafenib) and MEK inhibition (trametinib) as the main standard of treatment for advanced-stage BRAFV600E mutant melanomas, given their higher efficacy and longer time to relapse/resistance compared with single-agent BRAF inhibitors [43,50].
Although promising for BRAF V600E and K, there are very limited findings for BRAF inhibitors in melanomas harboring non-BRAF V600E mutations, which include BRAFV600R, BRAFV601K, and BRAF L597K [51,52]. Limited small individual trials showed that BRAF K601E and L597Q are at least partially sensitive to trametinib (MEK inhibitor), with 3 of 5 treated patients showing objective clinical and radiological response with a very manageable toxicity profile [51]. Similarly, another study including 45 patients with advanced/metastatic BRAF V600R mutant melanoma showed an overall response rate of 50% for the whole population, with a progression-free survival of 5.5 months [52]. Five objective responses were seen in 6 assessable patients with V600R BRAF mutation (total number of treated patients, n=9). The findings suggested that patients with BRAF V600R mutations can be treated successfully with oral BRAF inhibitors [52].
While melanoma classification is conventionally based on histological subtypes, classification based on molecular characteristics detected by novel sequencing techniques complements the histologic classification. This was highlighted in recent work by Siroy et al. Their study of 699 advanced-stage melanomas found significant associations of mutations with clinical subtypes and primary tumor location [53]. Indeed, the authors showed that BRAF V600E mutations were significantly associated with cutaneous and unknown primary melanomas (p<0.001), whereas KIT mutations were significantly associated with acral and mucosal melanomas (p<0.001) [53]. Additionally, BRAF V600E mutations were significantly associated with the trunk location compared with head and neck (p=0.0004), whereas TP53 mutations were significantly associated with head and neck compared with the trunk or extremities (p=0.03 and p<0.0001, respectively) [53].
The application of novel NGS techniques has enabled a better understanding of histogenesis in biphasic cutaneous tumors (cutaneous carcinosarcomas) as well as identification of potential therapeutic targets. Using laser microdissection and deep sequencing of the 2 different components of 6 cutaneous carcinosarcomas, Paniz-Mondolfi et al. showed that both elements of this infrequent tumor indeed share a common clonal origin [54]. In a similar case, Kiuru et al. showed an overlap in the mutations identified in both epithelial and sarcomatoid tumor compartments, confirming their clonal origin. Additionally, both components shared a truncating and a missense mutation in the patched gene (PTCH1). Both of these mutations are deleterious and can be targeted with smoothened (SMO) inhibitors [55].
Concluding key points
- Advances in molecular techniques have enabled development of numerous platforms with potential for application in the diagnosis and prognosis of challenging borderline and malignant melanocytic neoplasms as well as other dermatological malignancies.
- Our knowledge of the molecular landscape of these tumors will enable us to further elucidate different involved pathways that may harbor therapeutic targets. Also, it will enable us to identify resistance mechanisms, which would guide alternative therapeutic regimens.
- When implementing NGS-based platforms in clinical practice, considerations concerning bioinformatics infrastructure and facilities cost are of utmost importance.
*Helpful definitions
Sequencing methods:
Notice the 2 major sequencing methods mentioned in the text: sequencing by synthesis with dye termination (Illumina platforms) and sequencing by synthesis with ion semiconductor, which is based on the detection of hydrogen ions released during the polymerization of DNA (a.k.a. synthesis) (Thermofisher platforms). The released hydrogen leads to a change in the pH that is detected by an ion sensor, transforming it into an electrical signal that is captured. One of the limitations of the ion is homologous sequence homopolymer (i.e., GGGGGGGGG). These types of sequences lead to slippage and sequencing errors because such areas of the genome with homopolymeric sequences tend to have lower accuracy when sequenced using ion semiconductor methods. The new SOLiD platform by Thermofisher uses a universal primer and a set of fluorescent-labeled probes that are ligated to the target sequence (similar to the solid-phase method by Illumina) associated with periodic primer resetting, enabling repetitive sequencing of the same base and yielding a very high accuracy.
Accuracy: Accuracy in next-generation sequencing is measured using the Q score, also referred to as the Phred Quality score measure The higher the Q score, the higher is the base calling accuracy, i.e., the probability that the variant called on the sequencing is truly present and not a mere artifact. A Q score >30 is ideal, with a Q score >20 being acceptable (accuracies of 99.99% and 99%, respectively). Q scores are defined as a property that is logarithmically related to the base calling error probabilities (P): Q = −10 log10P.
Read Length: The read length refers to the length of the sequence generated after the library preparation. A library consists of the generated hybrids of target fragmented sequences (usually down to 150 bp) with the complementary barcoded sequences provided by the manufacturer (commonly referred to as “baits”). Once the library is ready, pair end sequencing (meaning sequencing from 5′ to 3′ and 3′ to 5′) occurs, simultaneously generating a bi-directional read for each sequence. Notice that the more fragmented the DNA is, the harder it is to sequence it, as the small fragments yield suboptimal reads for realignments. DNA fragmentation happens frequently with time. This is one of the reasons why relatively new or recent specimens perform better in NGS. This read length also varies with the different types of platforms.
Data Range: All sequencers produce FASTQ files. FASTQ format is a text-based format for storing both the biological sequence (usually nucleotide sequence) and its corresponding quality scores (Q scores). The size of these files containing sequencing data that is converted into multiple of the unit byte for digital information (megabytes, gigabytes, and even terabytes). The MiSeq platforms have relatively limited capacity in the depth of sequencing. Hence they generate less data per run compared with higher performance machines such as HiSeq. This is important to consider depending on the application and the gene panel tested. In common clinical practice when performing NGS for targeted therapies using a targeted exon panel, MiSeq platforms are efficient and provide a rapid turnover. In research-oriented facilities that perform deeper sequencing across a more expansive set of genes (400-500 or even whole-genome sequencing), HiSeq sequencing systems provide a higher quality and quantity of data.
Reference
- van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, et al. (2013) Whole-genome sequencing in health care. Recommendations of the European Society of Human Genetics. Eur J Hum Genet 21 (Suppl 1): S1-5. [Crossref]
- Jiang Z, Wang H, Michal JJ, Zhou X, Liu B, et al. (2016) Genome Wide Sampling Sequencing for SNP Genotyping: Methods, Challenges and Future Development. Int J Biol Sci 12: 100-108. [Crossref]
- Bastian BC1 (2002) Molecular cytogenetics as a diagnostic tool for typing melanocytic tumors. Recent Results Cancer Res 160: 92-99. [Crossref]
- Gerami P, Wass A, Mafee M, Fang Y, Pulitzer MP, et al. (2009) Fluorescence in situ hybridization for distinguishing nevoid melanomas from mitotically active nevi. Am J Surg Pathol 33: 1783-1788. [Crossref]
- Gerami P, Li G, Pouryazdanparast P, Blondin B, Beilfuss B, et al. (2012) A highly specific and discriminatory FISH assay for distinguishing between benign and malignant melanocytic neoplasms. Am J Surg Pathol 36: 808-817. [Crossref]
- Pouryazdanparast P, Cowen DP, Beilfuss BA, Haghighat Z, Guitart J, et al. (2012) Distinctive clinical and histologic features in cutaneous melanoma with copy number gains in 8q24. Am J Surg Pathol 36: 253-264. [Crossref]
- Gerami P, Scolyer RA, Xu X, Elder DE, Abraham RM, et al. (2013) Risk assessment for atypical spitzoid melanocytic neoplasms using FISH to identify chromosomal copy number aberrations. Am J Surg Pathol 37: 676-684. [Crossref]
- http://www.neogenomics.com/t_test/neosite-melanoma/: Neogenomics; 2016 [last accessed 02/10/2016].
- North JP, Garrido MC, Kolaitis NA, LeBoit PE, McCalmont TH, et al. (2014) Fluorescence in situ hybridization as an ancillary tool in the diagnosis of ambiguous melanocytic neoplasms: a review of 804 cases. Am J Surg Pathol 38: 824-831. [Crossref]
- http://www.genomics.agilent.com/en/CGH-SNP-Microarrays/Human-Genome-CGH-SNP-Microarrays 2016 [last accessed 02/16/2016]. [Crossref]
2021 Copyright OAT. All rights reserv
- http://www.affymetrix.com/estore/esearch/search.jsp?Ntt=ONCOSCAN&basic=1 last accessed 02/18/2016]. [Crossref]
- Bastian BC, Olshen AB, LeBoit PE, Pinkel D (2003) Classifying melanocytic tumors based on DNA copy number changes. Am J Pathol 163: 1765-1770. [Crossref]
- Chandler WM, Rowe LR, Florell SR, Jahromi MS, Schiffman JD et al. (2012) Differentiation of malignant melanoma from benign nevus using a novel genomic microarray with low specimen requirements. Arch Pathol Lab Med 136: 947-955. [Crossref]
- https://tcgadata.nci.nih.gov/tcga/tcgaCancerDetails.jsp?diseaseType=SKCM&diseaseName=Skin%20Cutaneous%20Melanoma 2016.
- Rock C, Clarke LE, Warf MB, Flake DD, Hartman AR, et al. (2014) Development and validation of a gene expression signature to distinguish malignant melanoma from benign nevi. Abstract. ASCO; May 201, Chicago, IL.
- Clarke LE, Warf MB, Flake DD 2nd, Hartman AR, Tahan S, et al. (2015) Clinical validation of a gene expression signature that differentiates benign nevi from malignant melanoma. J Cutan Pathol 42: 244-252. [Crossref]
- Loren E, Clarke EB, Kathryn A Kolquist, Colleen Rock (2015) A clinically validated gene expression score impacts diagnosis and management recommendations of melanocytic lesions by dermatopathologists. Abstract. The National Comprehensive Cancer Network , March 2015.
- Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74: 5463-5467. [Crossref]
- Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, et al. (1977) Nucleotide sequence of bacteriophage phi X174 DNA. Nature 265: 687-695. [Crossref]
- Schuster SC1 (2008) Next-generation sequencing transforms today's biology. Nat Methods 5: 16-18. [Crossref]
- Volmar KE, Idowu MO, Souers RJ, Nakhleh RE (2015) Molecular testing in anatomic pathology and adherence to guidelines: a College of American Pathologists Q-Probes Study of 2230 testing events reported by 26 institutions. Arch Pathol Lab Med 139: 1115-1124. [Crossref]
- Dummer R, Hauschild A, Lindenblatt N, Pentheroudakis G, et al. (2015) Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 26 Suppl 5:v126-132. [Crossref]
- Gabriel S, Ziaugra L, Tabbaa D (2009) SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet Chapter 2: Unit 2. [Crossref]
- Curry JL, Torres-Cabala CA, Tetzlaff MT, Bowman C, Prieto VG (2012) Molecular platforms utilized to detect BRAF V600E mutation in melanoma. Semin Cutan Med Surg 31: 267-273. [Crossref]
- Jiang Z, Rokhsar DS, Harland RM (2009) Old can be new again: HAPPY whole genome sequencing, mapping and assembly. Int J Biol Sci 5: 298-303. [Crossref]
- Jiang Z, Zhou X, Li R, Michal JJ, Zhang S, et al. (2015) Whole transcriptome analysis with sequencing: methods, challenges and potential solutions. Cell Mol Life Sci 72: 3425-3439. [Crossref]
- Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24: 133-141. [Crossref]
- http://www.illumina.com/ 2015 [cited 2016, last accessed 03/04/2016].
- http://www.thermofisher.com/us/en/home.html 2015 [cited 2016, last accessed 03/04/2016].
- Sethi N, MacLennan K, Wood HM, Rabbitts P1 (2016) Past and future impact of next-generation sequencing in head and neck cancer. Head Neck 38 Suppl 1: E2395-2402. [Crossref]
- http://cancergenome.nih.gov/abouttcga: U.S. Department of Health 2015[cited 2016, last accessed 03/04/2016].
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, et al. (2012) A landscape of driver mutations in melanoma. Cell 150: 251-263. [Crossref]
- Merid SK, Goranskaya D, Alexeyenko A (2014) Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC bioinformatics 15: 308. [Crossref]
- Sullivan R, LoRusso P, Boerner S, Dummer R1 (2015) Achievements and challenges of molecular targeted therapy in melanoma. Am Soc Clin Oncol Educ Book . [Crossref]
- Guan J, Gupta R, Filipp FV1 (2015) Cancer systems biology of TCGA SKCM: efficient detection of genomic drivers in melanoma. Sci Rep 5: 7857. [Crossref]
- Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247-269. [Crossref]
- Watson IR, Li L, Cabeceiras PK, Mahdavi M, Gutschner T, et al. (2014) The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res 74: 4845-4852. [Crossref]
- Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, et al. (2014) Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 5: 3116. [Crossref]
- Saito M, Shimada Y, Shiraishi K, Sakamoto H, Tsuta K, et al. (2015) Development of lung adenocarcinomas with exclusive dependence on oncogene fusions. Cancer Res 75: 2264-2271. [Crossref]
- Hutchinson KE, Lipson D, Stephens PJ, Otto G, Lehmann BD, et al. (2013) BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 19: 6696-6702. [Crossref]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, et al. (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367: 1694-1703. [Crossref]
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380: 358-365. [Crossref]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, et al. (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507-2516. [Crossref]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, et al. (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363: 809-819. [Crossref]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. (2015) Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 386: 444-451. [Crossref]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371: 1877-1888. [Crossref]
- Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, et al. (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371: 1867-1876. [Crossref]
- Johnson DB, Flaherty KT, Weber JS, Infante JR, Kim KB, et al. (2014) Combined BRAF (dabrafenib) and MEK inhibition (trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. Journal of Clinical Oncology 32: 3697-3704.
- Bowyer SE, Rao AD, Lyle M, Sandhu S, Long GV, et al. (2014) Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res 24: 504-508. [Crossref]
- Klein O, Clements A, Menzies AM, O'Toole S, Kefford RF, et al. (2013) BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer 49: 1073-1079. [Crossref]
- Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, et al. (2015) Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol 135: 508-515. [Crossref]
- Paniz-Mondolfi A, Singh R, Jour G, Mahmoodi M, Diwan AH, et al. (2014) Cutaneous carcinosarcoma: further insights into its mutational landscape through massive parallel genome sequencing. Virchows Arch 465: 339-350. [Crossref]
- Kiuru M, McDermott G, Coit DC, Berger MF, Busam KJ (2014) Basal cell carcinosarcoma with PTCH1 mutations in both epithelial and sarcomatoid primary tumor components and in the sarcomatoid metastasis. Am J Surg Pathol 38: 138-142. [Crossref]