Abstract
Cervical carcinoma is one of the highest causes of mortality in female and the number of patients is gradually increasing in the world. HeLa is a cervical carcinoma cell line and first established human immortalized cell line from cervical cancer patient. On the other hands, Heat shock transcription factor 1 (HSF1) is highly expressed in several cancer cells and tumors, and also involved in the malignancy. In order to investigate the role of HSF1 in cervical carcinoma, I established HeLa cell lines inducibly expressing constitutively active HSF1 (caHSF1) or dominant negative mutant RgHSF1, in a manner controllable by tetracycline. Expressed caHSF1 significantly suppressed HeLa cell growth, whereas RgHSF1 did not show appreciable effects. G1 arrest occurred in the HeLa cells expressing caHSF1, and cycline-dependent kinase (CDK) inhibitors p16 and p21 were up-regulated. To our knowledge, this is the first report that HSF1 may have a role to suppress the cancer cell growth. But importantly, this study suggests HSF1 may have more complicated roles than expected.
Key words
cervical carcinoma, caHSF1, HeLa, cell cycle, cell growth
Introduction
Cervical carcinoma incidence is increasing in incidence each year in the world, particularly among young women. It is widely known that human papillomaviruses (HPV) are significant risk factor for the development of cervical carcinoma [1,2]. HeLa is a cervical carcinoma cell line that has contributed to the various fields of research including cancer. HeLa is also the first established human immortalized cell line, and was originally obtained from a patient [3]. After the establishment of HeLa cell line, HeLa cells became indispensable for biological or medical science research at once and the contribution has been still going on. Importantly, it has been discovered that the p53 tumor suppressor pathway has been disrupted by HPV in these cell lines including HeLa and 90% of cervical carcinoma cells. In cervical carcinoma, p53 protein is known to be actively degraded by HPV E6 protein and thus stabilization and activation of p53 is suppressed. These reactions normally occur in response to HPV E7 oncogene oncoprotein expression [4-7].
Heat shock transcription factor (HSF1) is a well known transcription factor because it has an ability to induce a variety of famous chaperone protein called heat shock proteins (HSPs) against various stresses, and maintain the intracellular homeostasis and cell survival [8]. However, the analysis of prostate carcinoma cell lines PC-3 and its metastatic variant PC-3M revealed that HSF1 is highly expressed at least in some types of carcinoma for the first time [9]. Since this discovery, it has been widely accepted that HSF1 is highly expressed in various cancer cells, tissues, and tumors [9-12]. Similarly, increased expression of HSPs were reported in malignant fibrous histiocytoma, lymph node-negative breast cancer, melanoma, and node-positive breast carcinoma [13-16].
In this study, we show the novel function of HSF1 in cancer. Constitutively active HSF1 (caHSF1) [17] was observed to significantly suppresses the growth of cervical carcinoma HeLa cells. We also observed G1 arrest occurred in caHSF1 expressing HeLa cells and that CDK inhibitors p16 and p21 were also induced in these cells. Probably because of this mechanism, the growth suppression persisted for at least 30 days, as we discuss in the following (Figure 3B).
Materials and methods
Establishment of HeLa cells inducibly expressing caHSF1
To establish the HeLa cells with controllable expression of caHSF1, several different cells were created using various vectors in a stepwise manner described below. In the first step, expression vector for a tetR-VP16 fusion protein (pUHD15-1) [18] and the pcDNA3.1/Neo vector (Invitrogen) were co-transfected into HeLa cells, and the cell selecton was performed in the medium containing 1.5 mg/ml of G418 disulfate (Nacalai Tesque, Kyoto, Japan). The resultant cells were transiently transfected with the luciferase reporter plasmid [18], and some cell lines HeLa/tetVP2 expressing luciferase at a high level were successfully obtained by measuring the luciferase activity. cDNA for caHSF1 was amplified using PCR [17], and inserted into the pUHG10-3 vector (a gift from Dr. M. Gossen). The resulting vector was co-transfected with the pcDNA3.1/Hygro vector (Invitrogen) into the HeLa/tetVP2 cells, and the cells were incubated in medium containing 0.3 mg/ml of hygromycin (Nacalai Tesque) for 3-4 weeks with changing the medium every three days. Around 3-4 weeks after, several colonies appeared and each colony was independently picked up and defined as one single clone (one cell line), and named as caHSF1-1, caHSF1-2, … and caHSF1-100. After we expanded these cells, we performed Western blotting using extracts of the cells, and identified caHSF1 gene containing cell line (Figure 1A, upper). These HeLa cells did not express caHSF1 in the presence of tetracycline (2-5 μg/ml), and inducibly express caHSF1 by tetracycline withdrawal (when caHSF1 is not induced, these cells are called as ‘caHSF1-carrying cells’ in this manuscript). RgHSF1 is a mutant HSF1 containing dominant negative R71G mutation. The HeLa cells inducibly expressing RgHSF1 (called as ‘RgHSF1-carrying cells’ in this manuscript similar to caHSF1-carrying cells) were similary confirmed its expression by Western blot (Figure 1A, lower)
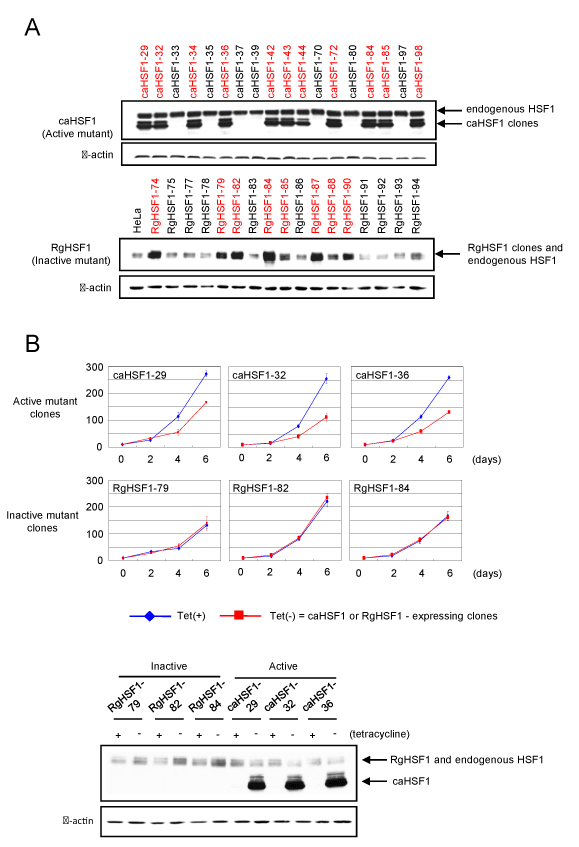
Figure 1. Confirmation of the establishment of HeLa cell lines inducibly expressing constitutively active HSF1 or dominant negative mutant HSF1 by Western blot and analysis of their growth for 6 days. (A) Establishment of HeLa cell lines inducibly expressing constitutively active HSF1 (caHSF1) (upper) or dominant negative mutant HSF1 (RgHSF1) (lower). RgHSF1-carrying cells have R71G mutation in HSF1 gene. (B) Cell growth analysis of three caHSF1 and RgHSF1 clones. Expression of RgHSF1 did not affect the cell growth, but caHSF1 expression prominently suppressed the growth of all caHSF1 clones (upper). Expression of caHSF1 and RgHSF1 was confirmed by Western blot (lower).
Cell culture
All cells were maintained in 37 degrees in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (inactivated 56 degrees for 30 min) and Penicillin-Streptomycin-Glutamine (100x) (GIBCO). caHSF1 positive cells were maintained in DMEM containing tetracycline (Sigma) at a concentration of 2-5µg/ml and 0.3 mg/ml of hygromycin (Nacalai Tesque) before experiments were performed. Under this condition, caHSF1 expression was inhibited. When caHSF1 was induced for experiments in the HSF1 positice cells, the cells were washed with PBS three times and add the DMEM described above without tetracycline. RgHSF1 was also induced in RgHSF1 positive cells using the same protocol.
Cell cycle analysis
The cell cycle was analyzed by FACS as described previously [19]. The cell were trypsinized and collected in 15 mL tube. Seventy percent ethanol was added drop by drop into the tube to fix the cells on ice. After adding 10 mL of seventy percent ethanol, fixation was kept O/N at 4 degrees. Prior to the FACS analysis, the cells were incubated with mouse anti-BrdU antibody (1:100 dilution, Pharmingen, CA) for 30 min. After incubation, centrifugation was performed and the pellets of these cells were resolved and washed three times with cold PBS. The pellets was resolved again and incubated with FITC-conjugated goat anti-mouse IgG antibody (1:100 dilution, Cappel) on ice keeping protection from light. Finally, the cells were completely resolved and mixed with 25 µg/ml propidium iodide (PI) before application and analyzed using a FACScan flow cytometer (Becton Dickinson, CA). Synthesized DNA was measured by the intensity of FITC, and cell survival was similarly measured by the intensity of PI.
Semi-Quantitative RT-PCR
The protocol for semi-quantitative RT-PCR was previously described [17]. The cells were washed three times with PBS and harvested with spatula. The PBS solution containing cells was collected into 1.5ml tube (Eppendorf) and centrifuged at 1,500 rpm for 5 min. After centrifugation, the supernatant was discarded and resultant cell pellet was immediately frozen with liquid nitrogen. Total RNA was extracted from the cells using TRIzol (Invitrogen) and typical RNA extracting reagents chloroform and ethanol. The resultant RNA pellets were dissolved in 100µl water and boiled for 5 min at 65 degrees. After 5 min, RNA-containing water was immediatedly moved to ice. After 5 min, the RNA concentration was measured. Two μg of RNA were applied for reverse-transcribed reaction by AMV kit (Invitrogen) using random primers. Synthesized cDNA was applied for PCR by Ex-Taq polymerase (Takara). As an internal control indicating the same amount of RNA was applied, the cDNA of ribosomal RNA S18 was also synthesized. Primer sequences were described previously [18], but all sequences are shown below again. For the detection of p16, p21 and S18, the following sequences of the primer sets were used: p16: 5’-CGCGGATCCGCCACCATGGAGCCGGCGGCG-3’ (forward primer) and 5-‘CGCG TTAACATCGGGGATGTCTGAGGG-3’ (reverse primer). P21: 5’-CGCGGATC CGCCACCA TGTCAGAACCGGCTGG-3’ (forward primr) and 5’-CGCGTTAACGGGCTTCCTCTT GGAGAAGATCAGC-3’ (reverse primer). S18: 5’-GGCAAGGAGCGATTTGCTGG-3’ (forward primer) and 5-‘GGGCTT ATCGGTAGGATTTCTGG-3’ (reverse primer). The gel for electrophoresis contained ethidium bromide. The amplified DNA was stained with ethidium bromide, and photographed using an Epi-Light UV FA1100 (Aisin Cosmos R&D Co., Japan), scanned, and and the quantities of the bands were determined by Image-J provided by NIH (http://imagej.nih.gov/ij/).
Western blot analysis
The cells were harvested using the same method for RT-PCR. The pellet of harvested cells were homogenized in NP40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP40) with protease inhibitors (1mM phenylmethylsulfomyl fluoride (PMSF), 1mg/ml pepstatin, 1mg/ml leupeptin) for 15 min on ice. Next, centrifugation was performed at 15,000 rpm for 10 min at 4 degrees, and the supernatant was collected. The protein concentration was measured by Bradford method. One hundred μg of soluble protein was applied to SDS-PAGE (10% acrylamide) for 2 hr at room temperature and blotted onto nitrocellulose membrane for 2 hr or O/N at 4 degrees. After blotting, the membrane was blocked with 5% skimmilk-containing PBS, and subjected to immunoblotting for O/N at 4°C using anti-HSF1j antiserum, and anti-β-actin antibody (Sigma). This membrane was washed with PBS three times and incubated with anti-rabbit goat IgG antibody (1:1000, for anti-HSF1j antiserum) or anti-mouse goat IgG antibody (1:1000, for anti-β-actin antibody) at room temperature for 1 hr. After incubaton, the membrane was washed with PBS three times and reacted with ECL Western blotting Detection Reagents (Amersham) for 2 min. In the last step, the membrane was exposed to X-ray film (HR-HA, FUJIFILM) for various minutes. Anti-HSF1j antiserum was raised by N.H. [19].
Statistical analysis
All data are shown with standard deviation. The significance was analyzed using student t test. When the p-value is less than 0.05, the difference was considered as significant.
Results
caHSF1 prominently suppressed the HeLa cell growth
In order to identify the roles of HSF1 in cervical carcinoma, we established HeLa cell lines inducibly expressing constitutively active HSF1 (caHSF1-carrying cells) (Figure 1A, upper panel). Similarly, HeLa cell lines inducibly expressing R71G mutation containing HSF1 (RgHSF1-carrying cells) were also established (Figure 1A, lower panel). We picked up three clones from each line and found the growth of caHSF1-carrying cells was prominently suppressed when caHSF1 was inducibly expressed by tetracycline withdrawal. The induction of RgHSF1 did not suppressed the growth of RgHSF1-carrying cells (Figure 1B). This result indicates caHSF1 has a specific and indispensable function for the growth suppression that other proteins does not have.
caHSF1 affects HeLa cell cycle
In order to examine the mechanism by which the caHSF1 expression suppressed HeLa cell growth, we performed the FACS-based analysis of cell cycle at 0 and 4th day after caHSF1 induction. The G1 proportion was increased by 54.5%, indicating G1 arrest. The G2/M proportion was prominently decreased at 4th day after caHSF1 was induced (Figure 2A and 2B). We also tried to examine at later stages for example 18th or 30th day, however, we could not obtain enough number of cells because caHSF1-expressing cells did not proliferate at all. Similarly, we could not perform the FACS analysis at later stages. As Figure 2A and 2B indicate, caHSF1 altered the HeLa cell cycle and caused G1 arrest.
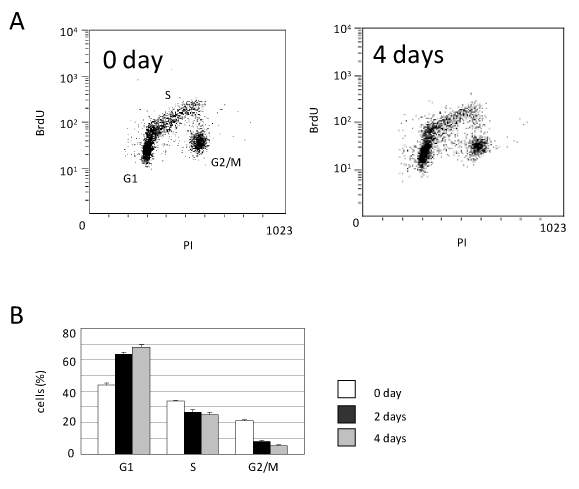
Figure 2. Cell cycle analysis using FACS. (A) The picture of the distribution of caHSF1-expressing HeLa cells (clone caHSF1-32) at 0 and 4th day after induction of caHSF1 by tetracycline withdrawal. (B) The proportion of the cells of each cell cycle phase during 4 days. At 4th day, the cells at G1 phase increased but the cells at G2/M phase prominently decreased.
CDK inhibitors are induced by HSF1
Because the cell cycle was modified and G1 arrest was observed, we examined the expressions of the genes for CDK inhibitors by RT-PCR. we found induction and increased expression of p16 and p21 (Figure 3A). In the last experiment, the cell growth of caHSF1-expressing cells and RgHSF1-expressing cells was examined for 30 days. In RgHSF1 -carrying cells, the growth was not changed regardless of RgHSF1 expression. In contrast, the growth of caHSF1-carrying cells when caHSF1 inducibly expressed were siginificantly suppressed for 30 days. The same cells showed normal cell growth similar to RgHSF1-carrying cells and RgHSF1-expressing cells when caHSF1 expression was inhibited by tetracycline. However, after caHSF1 was inducibly expressed until 6th day and re-suppression of its expression started by tetracycline addition on the 7th day, the cell growth was recovered and became normal (Figure 3B). These data indicate that caHSF1 induces expressions of CDK inhibitors p16 and p21 and suggest that this induction causes G1 arrest and growth retardation in cervical carcinoma HeLa cells.
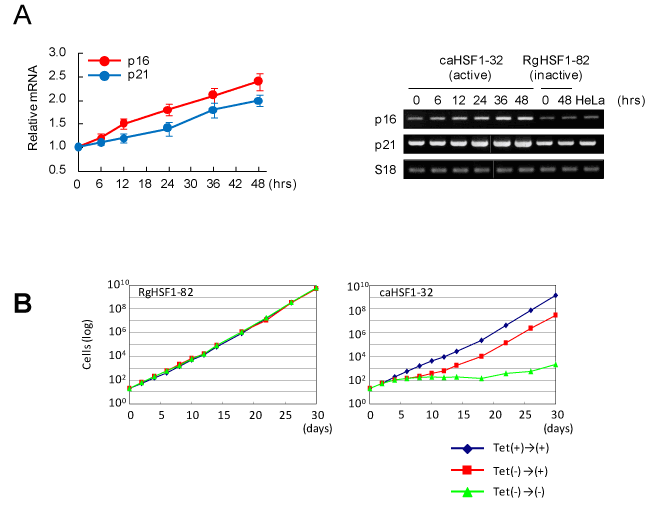
Figure 3. CDK inhibitors p16 and p21 are induced in the HeLa cell line inducibly expressing caHSF1. (A) Increased expression of p16 and p21 transcripts after caHSF1 was induced by tetracycline withdrawal analyzed by RT-PCR. Time-development for 48hrs (left) and the picture of the bands of p16, p21, and -actin amplified by PCR. (B) The growth curves of RgHSF1-carrying (clone RgHSF1-82) and caHSF1-carrying (clone caHSF1-32) cells. When both HSF1 expression was inhibited, the cells showed normal growth (blue). In RgHSF1-carrying cells, the cells showed normal growth again regardless of whether RgHSF1 was induced or not (green) (left). In caHSF1-carrying cells, they showed prominently retarded growth for 30 days (green) when caHSF1 was induced and expressed. When caHSF1 was expressed for 6 days and it was inhibited at 7th day, the growth was retarded until 12th day but recovered normal growth (red).
Discussion
In this paper, we showed constitutively active HSF1 (caHSF1) prominently suppresses the growth of HeLa cells. This finding is very surprising, because it stands in opposition to the results of our previous experiments and other papers indicating that cancer cell growth is prominently suppressed by HSF1 knockdown, not overexpression or activation [10,11]. In 2007, Dai et al. 2007 showed that HSF1 knockdown effects on viable cell percentage of various cancer cells including breast cancer MCF-7, prostate cancer PC-3, kidney cancer HEK293 and cervical cancer HeLa [10]. Their data showed that the cell viability was significantly reduced by HSF1 knockdown in all cancer cells including HeLa cell. Rossi et al. performed HSF1 knockdown in HeLa cells and examined their sensitivity to anti-cancer drug cisplatin, but the sensitivity and appearance of apoptotic HeLa cells were not affected by HSF1 knockdown. In the latter study, the growth of HSF1-knockdown HeLa cells was not examined [22].
Mendillo et al. showed high expression of HSF1 in tumors in vivo by immunohistochemistry [23]. HSF1 was positive in prostate, colon, lung, pancreas, meningioma, and cervix tumors, and strongly positive in nucleus of these tumors. These data indicate HSF1 is highly expressed in cancer cells and tumors, and that this is indispensable for cancar cell growth but not for the growth of normal cells.
On the other hand, why and how does caHSF1 inhibit the growth of the cervical carcinoma HeLa cells ? As the answer to the ‘how’, we discovered that p16 and p21 are induced by caHSF1 for the first time, to our knowledge. But needless to say, the induction of p16 and p21 explain just a part of the mechanism for this phenomenon. HSF1 induces a diverse set of genes including pro-apoptotic gene TDAG51 (24), immune system and cell cycle regulating transcription factor NFATc2 [17,25-28], and a various kind of genes [17]. Other HSF1 target genes must be involved in the growth inhibition of cervical carcinoma HeLa cells.
Concerning the growth inhibition of cervical carcinoma cells, many papers have been published and some of them clearly showed the cellular senescence of these cells including HeLa [29-34]. The immortalization and transformation occurred in cervical carcinoma is caused by the E6 and E7 genes of human papillomaviruses (HPV) [35,36], and frequent loss of E2 gene is also indispensable for this immortalization and transformation [37]. Thus, introduction of normal E2 gene to the cervical carcinoma cells inhibits their growth and these cells showed senescence-associated β-galactosidase activity (SA-β-Gal) indicating cellular senescence. E8^E2C proteins (encoded by the same E2 gene) induced G1 arrest with higher efficiency than E2 and also caused cellular senescence identified by SA-β-Gal expression in HeLa cells [29,33].
In this study, we showed the caHSF1 causes cell growth inhibition in cervical carcinoma HeLa cells similar to the melanoma cell growth inhibition caused by HSF1 knockdown in our previous study [11]. Whether caHSF1 can cause growth inhibition in other cancer cells as well as in HeLa is unknown, and it is not straightforward to address this question. For example, HeLa cells are aneuploidy and have higher rates of mis-segregation, 0.24% per mitosis for chromosome 8 and 0.39% for chromosome 12 [38]. Thus, there might be a possibility that caHSF1 cannot always inihibit the growth of cervical carcinoma cells including HeLa because the instability of their genome might modify the mechanism of cell growth regulation by HSF1 in several cervical carcinoma cell lines and many HeLa sub-lines.
Conflict of interest
The author declares I have no conflict of interest.
Acknowledgements
This work was supported by Nipponham Foundation for the Future of Food (2016-B-3 N.H.), JSPS KAKENHI Grant-in-Aid for Young Scientist (B) (23700512 N.H.), Grant-in-Aid for Scientific Research (C) (25430090 N.H.), and Takeda Science Foundation (23009 N.H.). Thanks to Drs. Shigeru Kakuta, Ken-ichiro Otsuyama, Takeshi Honda, Hiromi Yamane, Tayoko Ishimura, Rumi Kitamura, Takao Kitagawa, Shusaku Uchida, Yoshifumi Watanabe, Hideshi Yokoyama, and Shuji Noguchi for encouragement and helps in experiments. I would like to thank to Yamaguchi University Center for Gene Research, Dr. Yo-ichi Mizukami and Center Staffs for the support of my research. Special thanks to Drs. Sayo Shoda, Masayuki Nakamura, Yasuhiro Kuramitsu, Shohei Fujimoto, Eisuke Enoki, and Yoshiharu Kamosaki for research assistance and encouragement. I also thank to the students of Yamaguchi Kojo School of Nursing and Yamaguchi University School of Medicine.
References
- Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, et al. (2005) Carcinogenicity of human papillomaviruses. Lancet Oncol 6: 204. [Crossref]
- Schiffman MH, Brinton LA (1995) The epidemiology of cervical carcinogenesis. Cancer 76: 1888-1901. [Crossref]
- Masters JR (2002) HeLa cells 50 years on: the good, the bad and the ugly. Nat Rev Cancer 2: 315-319. [Crossref]
- Münger K, Scheffner M, Huibregtse JM, Howley PM (1992) Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv 12: 197-217. [Crossref]
- Narisawa-Saito M, Kiyono T (2007) Basic mechanisms of high-risk human papillomavirus- induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci 98: 1505-1511. [Crossref]
- Tewari KS, Monk BJ (2014) New strategies in advanced cervical cancer: from angiogenesis blockade to immunotherapy. Clin Cancer Res 20: 5349-5358. [Crossref]
2021 Copyright OAT. All rights reserv
- Sheaffer AK, Lee MS, Qi H, Chaniewski S, Zheng X, et al. (2016) A Small Molecule Inhibitor Selectively Induces Apoptosis in Cells Transformed by High Risk Human Papilloma Viruses. PLoS One 11: e0155909. [Crossref]
- Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319, 916-919. [Crossref]
- Hoang AT, Huang J, Rudra-Ganguly N, Zheng J, Powell WC, et al. (2000) A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am J Pathol 156: 857-864. [Crossref]
- Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130: 1005-1018. [Crossref]
- Nakamura Y, Fujimoto M, Hayashida N, Takii R, Nakai A, et al. (2010) Silencing HSF1 by short hairpin RNA decreases cell proliferation and enhances sensitivity to hyperthermia in human melanoma cell lines. J Dermatol Sci 60: 187-192. [Crossref]
- Nakamura Y, Fujimoto M, Fukushima S, Nakamura A, Hayashida N, et al. (2014) Heat shock factor 1 is required for migration and invasion of human melanoma in vitro and in vivo. Cancer Lett 354: 329-335. [Crossref]
- Têtu B, Lacasse B, Bouchard HL, Lagacé R, Huot J, et al. (1992) Prognostic influence of HSP-27 expression in malignant fibrous histiocytoma: a clinicopathological and immunohistochemical study. Cancer Res 52: 2325-2328. [Crossref]
- Ciocca DR, Clark GM, Tandon AK, Fuqua SA, Welch WJ, et al. (1993) Heat shock protein hsp70 in patients with axillary lymph node-negative breast cancer: prognostic implications. J Natl Cancer Inst 85: 570-574. [Crossref]
- Protti MP, Heltai S, Bellone M, Ferrarini M, Manfredi AA, et al. (1994) Constitutive expression of the heat shock protein 72 kDa in human melanoma cells. Cancer Lett 85: 211-216. [Crossref]
- Têtu B, Brisson J, Landry J, Huot J (1995) Prognostic significance of heat-shock protein-27 in node-positive breast carcinoma: an immunohistochemical study. Breast Cancer Res Treat 36: 93-97.
- Hayashida N, Fujimoto M, Tan K, Prakasam R, Shinkawa T, et al. (2010) Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J 29: 3459-3469. [Crossref]
- Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89: 5547-5551. [Crossref]
- Shinkawa T, Tan K, Fujimoto M, Hayashida N, et al. (2011) Heat shock factor 2 is required for maintaining proteostasis against febrile-range thermal stress and polyglutamine aggregation. Mol Biol Cell 19, 3571-3583. [Crossref]
- Hayashida N (2015) Non-Hsp genes are essential for HSF1-mediated maintenance of whole body homeostasis. Exp Anim 64: 397-406. [Crossref]
- Hayashida N (2015) Set1/MLL complex is indispensable for the transcriptional ability of heat shock transcription factor 2. Biochem Biophys Res Commun 467: 805-812. [Crossref]
- Rossi A, Ciafrè S, Balsamo M, Pierimarchi P, Santoro MG (2006) Targeting the heat shock factor 1 by RNA interference: a potent tool to enhance hyperthermochemotherapy efficacy in cervical cancer. Cancer Res 66: 7678-7685. [Crossref]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, et al. (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150: 549-562. [Crossref]
- Hayashida N, Inouye S, Fujimoto M, Tanaka Y, Izu H, et al. (2006) A novel HSF1-mediated death pathway that is suppressed by heat shock proteins. EMBO J 25: 4773-4783. [Crossref]
- Viola JP, Kiani A, Bozza PT, Rao A (1998) Regulation of allergic inflammation and eosinophil recruitment in mice lacking the transcription factor NFAT1: role of interleukin-4 (IL-4) and IL-5. Blood 91: 2223-2230. [Crossref]
- Baksh S, Widlund HR, Frazer-Abel AA, Du J, Fosmire S, et al. (2002) NFATc2-mediated repression of cyclin-dependent kinase 4 expression. Mol Cell 10: 1071-1081. [Crossref]
- Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, Tertilt C, et al. (2006) Specific and redundant roles for NFAT transcription factors in the expression of mast cell-derived cytokines. J Immunol 177: 6667-6674. [Crossref]
- Karwot R, Maxeiner JH, Schmitt S, Scholtes P, Hausding M, et al. (2008) Protective role of nuclear factor of activated T cells 2 in CD8+ long-lived memory T cells in an allergy model. J Allergy Clin Immunol 121: 992-999. [Crossref]
- Hwang ES, Riese DJ 2nd, Settleman J, Nilson LA, Honig J, et al. (1993) Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J Virol 67: 3720-3729. [Crossref]
- Goodwin EC, Yang E, Lee CJ, Lee HW, DiMaio D, et al. (2000) Rapid induction of senescence in human cervical carcinoma cells. Proc Natl Acad Sci U S A 97: 10978-10983. [Crossref]
- DeFilippis RA, Goodwin EC, Wu L, DiMaio D (2003) Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol 77: 1551-1563. [Crossref]
- 32. Johung K, Goodwin EC, DiMaio D (2007) Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J Virol 81: 2102-2116.
- Fertey J, Hurst J, Straub E, Schenker A, Iftner T et al. (2011) Growth inhibition of HeLa cells is a conserved feature of high-risk human papillomavirus E8^E2C proteins and can also be achieved by an artificial repressor protein. J Virol 85: 2918-2926. [Crossref]
- Maritz MF, van der Watt PJ, Holderness N, Birrer MJ, Leaner VD (2011) Inhibition of AP-1 suppresses cervical cancer cell proliferation and is associated with p21 expression. Biol Chem 392: 439-448. [Crossref]
- Pirisi L, Creek KE, Doniger J, DiPaolo JA (1988) Continuous cell lines with altered growth and differentiation properties originate after transfection of human keratinocytes with human papillomavirus type 16 DNA. Carcinogenesis 9: 1573-1579. [Crossref]
- DiMaio D (1991) Transforming activity of bovine and human papillomaviruses in cultured cells. Adv Cancer Res 56: 133-159. [Crossref]
- Romanczuk H, Howley PM (1992) Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci USA 89: 3159-3163. [Crossref]
- Shi Q, King RW (2005) Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 437: 1038-1042. [Crossref]