Abstract
We aimed to report a rare case with torpedo maculopathy (TM). 61 years old manadmitted to our clinic with complain of the loss of central vision in his left eye for two months. Ocular and systemic medical history was unremarkable. His visual acuities were 20/20 in the right eye and 14/20 in the left eye without any pathology in the biomicroscopic examination. Fundusexamination revealed a flat, hypopigmented, fusiform chorioretinalles ion with well-defined margins located at the temporal sector of the macular regionand next to fovea in the right eyeandfoveal retina pigment epithelium (RPE) alterations in the left eye. TMwas diagnosed basedon the characteristic view and the location of the lesion. As TM is hypopigmented congenital nevus of theRPE, it should be differentiated fromsome pigment-related disorders such as choroidalmelanoma and nevus, congenital hypertrophy of RPE and the congenital pigmented lesions havingthe potential of malignancy as in Gardner’s syndrome.
Key words
retina pigment epithelium, torpedo maculopathy, hypopigmented nevus
Introduction
Torpedo maculopathy (TM), a rare benign disorder is characterized by hypopigmented congenital nevus of retina pigment epithelium (RPE) located along the horizontal raphe without significant vision loss[1,2]. Its synonyms are solitary hypopigmented nevus,amelanotic nevus, hypomelanoticor albinotic nevi of RPE, solitary amelanotic spot, congenital hypomelanotic freckle, paramacularalbinotic spot syndrome and paramacularcoloboma [1-8].As there is no sufficient data on the epidemiology of TM, it has not been known that its incidence and demographic properties such as gender and race. Here, we report a case with TM because it is a rare disorder.
Case report
A 61 years old male admitted to our university hospital with the complaint of central vison loss in her left eye for a period of two months. He had no remarkable medical history of any systemic and ocular disease. On initial examination, his corrected visual acuity was 20/20 in the right eye and 14/20 in the left eye. The intraocular pressure was 14 mmHg in both eyes. Direct and indirect light reflexes were intact, without a relative afferent pupillary defect. The anterior segment examination and extraocular movements were normal. Fundus examination revealed a flat, hypopigmented, fusiform chorioretinal lesion with well-defined marginsand a tip pointing toward the fovea located at the temporal portion of the macular region and next to fovea in the right eyeand foveal RPE alterations in the left eye (Figure 1A, B).The lesion had dimensions of horizontally 2.43 mm X vertically 1.23 mm and involved the temporal half of fovea. Humphrey Visual field perimetry showed a small paracentral scotoma corresponding to the temporal portion of the macular lesion in the right eye.Fundus fluorescein angiogram (FFA) showed hypofluorescence at nasal one third part of lesion and hyperfluorescence at temporal two third part of lesion, located at the temporal half of fovea in the right eye. Late stage angiograms revealed no any changes in the same eye (Figure 1C,E). The FFA of the left eye showed focal RPE window defects in the superonasal to optic disc (Figure 1D, F).Optic coherence tomography (OCT) revealed choroidalhyperreflectivity andthinningofRPE at whole lesion region andchoroidal excavation in temporal half of the lesion in the right eye while OCT revealed parafoveal mild thinning of retina (Figure 2). Torpedo maculopathy was diagnosed based on the characteristic view and the location of the lesion. In systemic examinations, no pathological finding was obtained. The neurology consultation and computerized brain tomography for scotomas did not any reveal pathological finding.
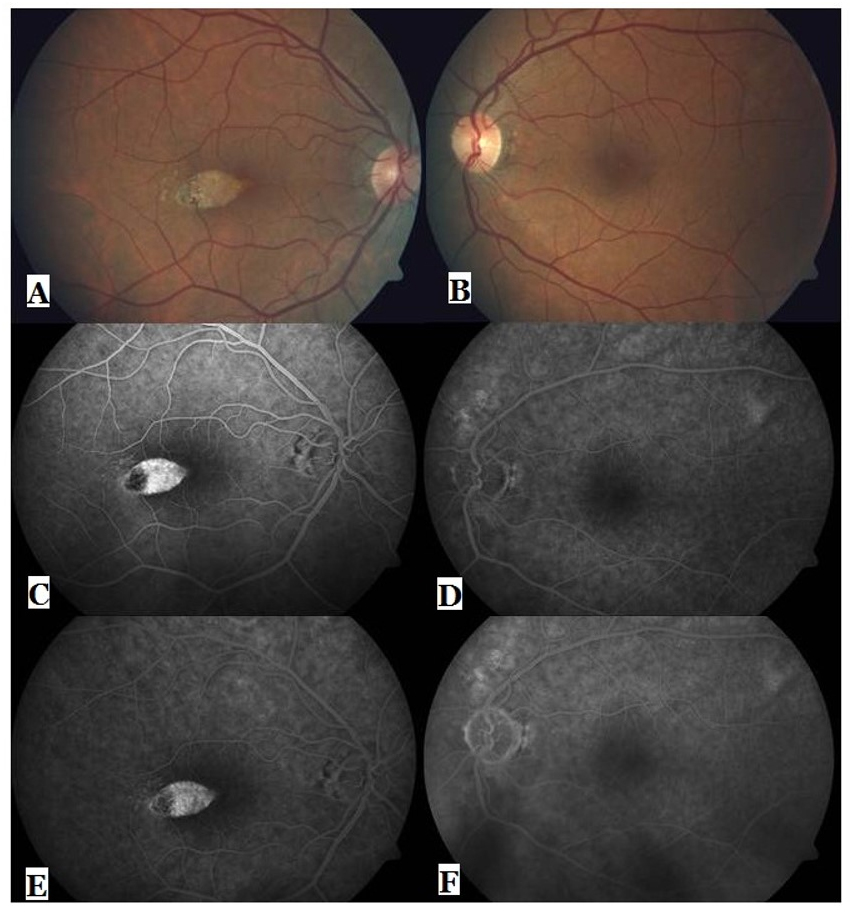
Figure 1. Color fundus images (A, B), early (C,E) and late phase (D, F) angiograms of the both eyes.
Conclusion
Torpedo maculopathy is firstly described by Roseman and Gassas a focal unilateral congenital abnormality of the RPE[1]. It is usually detected an accidental finding. It seems as hypopigmented macular lesion with well-defined margins and a torpedo-like tip pointing toward the foveola at the temporal portion of maculain single focus in single eye [1-8]. Patients withTMare asymptomatic, and have normal visual acuity. However, due to photoreceptor loss at the regionofmacular lesion, paracentralscotoma on visual field perimetry, corresponding to the localization of thelesion may be detected [2,8,11,12]. In our case, Humphrey Visual field perimetry showed aparacentral scotoma corresponding to the temporal portion of the macular lesion in the right eye. Interestingly, our case had complain of the loss of central vision in his left eye but not right eye whileas TMwas in the right eye. So, we accidentally detected the lesion in the right eye.
Although the etiology of TMis unknown, some investigators have several hypotheses on this disorder. Pian et al. have hypothesized that itis a developmental defect in the nerve fiber layer at the horizontal raphe due to the differentiation deficiency of the ocular tissuesin the postnatal period [5]. Golchet et al. have considered that it results from a defect in the normal vascular development of choroid [2]. Shields et al. have speculated thatit might be related to thecongenital persistent defect in RPE embryogenesis or development defect in the fetal temporal bulgebecause of the uniform location and size of the lesion [6].
The diagnosis of this disorder is clinically made on the basis of characteristic torpedo-like shape and location of the lesion [1-7]. However, multimodal imaging including FFA, OCT, and fundus autofluorescence might also be useful for diagnosis and differential diagnosis. The FFAcan reveala transmission hyperfluorescence or RPE window defect in lesion region[2,4,8]. In FFA, we observed a transmission hyperfluorescence at nasal two third part of lesion and hypofluorescence at temporal one third part of lesion, without any changes in the late phases in the right eye. This hypofluorescence may be related the region of choroidal excavation or choroidalhyperreflectivity in OCT.OCT can reveal retinal thinning, increased choroidal reflectance or RPE hyporeflectivity, and shallow serous neurosens orial detachment and subretinal cleft [9-12]. In a recent study on OCT findings intorpedo maculopathy, two patterns of abnormality as type 1 and 2 were identified Wong et al. Type 1 has normal inner retina, attenuation of interdigitation zone and ellipsoid zone in outer retina, without outer retinal cavitation. Type 2 has normal inner retina, thinning of outer nuclear layer, loss of interdigitation zone and ellipsoid zone in outer retina and outer retinal cavitation +/- inner choroidal excavation [13]. We had detected choroidalexcavation, hyperreflectivity and thinning of outer nuclear layer, loss of interdigitation zone and ellipsoid zone in temporal portion of the lesion and RPE thinning in whole lesion region in OCT scans. So, our case might be considered type 2 torpedo maculopathy according to OCT classification defined by Wong et al. [13].
AlthoughTMis a benign congenital nevus of the RPE, it should be differentiated fromsome pigment-related disorders such as choroidal melanoma and nevus, congenital hypertrophy of RPE (CHRPE) and pigmentary retinal disorders associated with familial poliposis syndromes such as especially Gardner’s syndrome because of their potential of the malignancy. The RPE lesions in Gardner’s syndrome are usually bilateral, smaller, irregularly shaped, solitary and multitidous located out of the macula [14-16]. Additionally, it can be differentiated from CHRPE with its characteristic aspect as fusiform or torpedo-like shape. The other differential diagnosis for this lesion includes congenital albinotic spots, trauma, drugs, infective or inflammatory chorioretinal scars and atypical Best's disease. The lesions in these conditions have highly irregular shape and variable location [14-16].
Authorship and contribution
All authors listed on the title page made significant contributions to this manuscript.
Acknowledgements
None
Funding
No funding was received for this study.
Competing interest
The authors have no conflict of interest or financial relationships related to this manuscript.
References
2021 Copyright OAT. All rights reserv
- Roseman RL, Gass JDM (1992) Solitary hypopigmented nevus of the retinal pigment epithelium in the macula. Arch Ophthalmol 110: 1358–1359. [Crossref]
- Golchet PR, Jampol LM, Mathura JR, Daily MJ (2010) Torpedo maculopathy. Br J Ophthalmol94: 302–306.
- Daily MJ (1993) Torpedo maculopathy or paramacular spot syndrome. In: New Dimensions in Retina: Nov. 10-13, Chicago.
- Barbazetto IA, Maris PJ Jr, Greenstein VC (2008) Solitary albinotic spot of the retinal pigment epithelium: a functional and imaging study. Klin Monbl Augenheilkd 225: 295-297. [Crossref]
- Pian D, Ferrucci S, Anderson S, Wu C (2003) Paramacularcoloboma. Optom Vis Sci 80: 556–563. [Crossref]
- Shields CL, Guzman JM, Shapiro MJ, Fogel LE, Shields JA (2010) Torpedo maculopathy at the site of the fetal ‘bulge’. Arch Ophthalmol 128: 499–501. [Crossref]
- Rigotti M, Babighian S, Carcereri De Prati E, MarchiniG (2002). Three cases of a rare congenital abnormality of the retinal pigment epithelium: torpedo maculopathy. Ophthalmologica 216: 226–227. [Crossref]
- Pilotto E, Zannin ME, Convento E, Cortese M, MidenaE (2013) Torpedo maculopathy: a morphofunctional evaluation. Int Ophthalmol 33: 71–74. [Crossref]
- Su Y, GurwoodAS (2010) Neurosensory retinal detachment secondary to torpedo maculopathy.Optometry 81:405–407. [Crossref]
- Sanabria MR, Coco RM, SanchidrianM (2008) OCT findings in torpedo maculopathy. Retin Cases Brief Rep 2: 109–111. [Crossref]
- Tsang T, Messner LV, Pilon A, Lombardi L (2009) Torpedo maculopathy: in-vivo histology using optical coherence tomography. Optom Vis Sci8 6: E1380–1385. [Crossref]
- Shields C, Materin MA, Walker C, Marr BP, Shields JA (2006) Photoreceptor loss overlying congenital hypertrophy of the retinal pigment epithelium by optical coherence tomography. Ophthalmology 113: 661–665. [Crossref]
- Wong EN, Fraser-Bell S, Hunyor AP, Chen FK (2015) Novel optical coherence tomography classification of torpedo maculopathy. Clin Experiment Ophthalmol 4 3: 342-348 .[Crossref]
- Tiret A, Taiel-Sartral M, Tiret E, LarocheL (1997) Diagnostic value of fundus examination in familial adenomatous polyposis. Br J Ophthalmoln 81: 755–758. [Crossref]
- Streeten BW (1969) Development of the human retinal pigment epithelium and the posterior segment. Arch Ophthalmol 8 1:383-394. [Crossref]
- Shields JA, Shields CL (2008) Tumours and related lesions of the pigment epithelium. In: Shields JA, Shields CL., eds. Intraoculartumours: an atlas and textbook. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 431-480.