A natural compound Resveratrol (Rsv), which is found in high concentrations in grape skins and red wine, is expected to be a leading drug for elongating the life span and preventing oncogenesis. This compound is reported to have various beneficial effects on health, including the activation of complex I and anti-oxidative stress. We have been focusing on the regulation of the expression of human genes and have analyzed the promoter activities of several genes that encode DNA repair-function-associated protein factors. Notably, the 5’-upstream regions of these genes very frequently contain the duplicated GGAA-motifs that allow HeLa S3 cells to respond to Rsv. In this review, we discuss the molecular mechanism by which Rsv regulates the expression of genes associated with DNA repair. We suggest the possibility of developing new anti-aging/cancer drugs without harmful side effects, based on a new concept of ameliorating or maintaining the cellular NAD+/NADH level.
ETS, GGAA, mitochondria, NAD+, NADH, Resveratrol, telomere, TP53
Abbreviations: CoQ: Coenzyme Q; CR: calorie restriction; 2-DG: 2-deoxy-D-glucose; HELB: helicase B; IFN: interferon; Luc: luciferase; mtDNA: mitochondrial DNA; NAD: nicotinamide adenine dinucleotide; OXPHOS: oxidative phosphorylation; Rsv: trans-resveratrol; TPA: 12-O-tetradecanoyl-phorbol-13-acetate; TE: transposable element; TF: transcription factor; UPRmt: mitochondrial unfolded protein response
To date, various DNA-repair associated protein factors have been identified and studied. These include DNA polymerases, DNA helicases, and cell cycle regulating proteins. Some of the DNA polymerases are essential for the repair synthesis in damaged parts of the genome [1]. DNA helicases are DNA-binding proteins that have various functions such as unwinding DNA double-strands and changing chromatin structures [2,3]. Several of the cell cycle regulating proteins control the progression of G1/S and play important roles as tumor suppressors, such as the p53[4,5] and RB [6] proteins, which are encoded by the TP53 and RB1 genes, respectively. These DNA repair regulatory factors are thought to control cellular senescence, which is suggested to be linked to the maintenance of telomeres [7,8]. For example, mutations of the WRN gene, which encodes a Rec Q helicase, are known to cause Werner’s syndrome [9]. We previously observed that the expression of the human TP53 and WRN genes (and their promoter activities) is induced in HeLa S3 cells by treatment with trans-Resveratrol (Rsv) [10,11], which is a natural compound that is expected to elongate the healthy life span [12,13]. Moreover, Rsv upregulates the expression of the HELB gene [10,14], which encodes DNA replication and DNA double strand break repair-helicase HELB (HDHB) [15-18]. Taken together, these observations imply that DNA-repair associated- and DNA/damage response-protein encoding genes are simultaneously regulated, at the transcriptional levels, by Rsv.
In general, the DNA repair and the genome maintenance system are thought to be tightly linked with the cellular aging process and oncogenesis [19, 20]. A recent analysis of the human cancer genome revealed a number of cancer-driver genes, including TP53 and RB1 [21,22]. The incidence of cancer in humans increases exponentially with age, suggesting that aging is the strongest demographic risk factor for most human malignancies [23,24]. The investigation of the molecular mechanisms involved in the genome maintenance system, which is affected by Rsv—a natural compound that has health benefits—will be very important in the development of anti-aging/cancer drugs. In this review article, we re-examine the 5’-flanking regions of the human genes that encode the DNA-repair factors and discuss the mechanisms underlying the response to Rsv.
Transcription is the fundamental biological process through which mRNAs are synthesized for translation into proteins. In eukaryotic cells, the formation of a pre-initiation complex, recruiting RNA polymerase II (RNA pol II) near a TATA-box, is believed to be the most essential factor for the initiation of transcription [25]. However, it is estimated that TATA-less promoters account for 76% of the upstream region of human genes [26]. We reported that 81% of the promoters, which contain no obvious consensus TATA-box sequences, have at least one duplicated 14-bp with GGAA (TTCC) core motif within the 500-bp upstream region [27]. The GGAA motif-containing sequences are recognized and bound by the ETS family proteins and other transcription factors (TFs) [28-30]. For example, it has been shown that ETS family protein ELF-1 binds to the duplicated GGAA motifs to regulate the expression of the antiviral OAS1, which belongs to the interferon-stimulated genes (ISGs) [31]. Recently, Chuong et al. showed—using a ChIP-seq analysis—that STAT1 binds to the interferon-g (IFNG) activated sequences (GAS), 5’-TTCCGGGAA-3’ and 5’-TTCCTGGAA-3’, in the MER41B LTR consensus sequence in IFNG-stimulated HeLa cells, suggesting that STAT1 plays an important role in the innate immunological responses [32]. The results were very interesting as they imply that ancestral retroviruses helped to confer the activation system in which transcription is activated upon IFN stimulation. Moreover, interferon regulatory factors (IRFs) can also recognize and bind to the interferon-stimulated response element (ISRE) or the GAS [33]. A recent study showed that the antiviral innate immune response, which is mediated by the TFAM-cGAS-STING-IRF3 signaling pathway, is primarily caused by mitochondrial DNA (mtDNA) stress through viral infection [34]. These observations suggest that the STAT and IRF protein factors are involved in transcriptional regulation through the duplicated GGAA (TTCC) motifs. The competitive binding of different GGAA-motif binding proteins to the same or the adjacent GGAA sequence means that the genetic expression is suitably adjusted by the expression profile of the GGAA-binding proteins in the cells [27]. Further surveillance of the 5’-upstream regions of DNA repair-, interferon response-, and mitochondrial function-associated genes revealed that their expression regulatory regions contain duplicated GGAA motifs near the transcription start sites [35-37]. The observations imply that some of the essential characteristics of cancer, including the DNA damage response and mutations, inflammation, and failure in the mitochondrial respiratory system, could be caused by changes in the profile of the GGAA (TTCC)-binding TFs [38]. Of note, GABP (NRF2), which belongs to the ETS family, is required for mitochondrial biogenesis [39]. A number of cancer causative stresses, including DNA damage responses, are thought to be tightly linked with the aging process [19, 40, 41]. Thus, the establishment of a method to prevent cancer with the use of drugs or gene expression enhancing/inhibiting vectors would also have clinical application in the prevention of aging-related diseases.
It was previously shown that the promoter activities of the human telomere maintenance factor-encoding genes positively respond to Rsv [10,42]. Moreover, our recent study showed that the expression of the human TP53 and HELB genes is upregulated by Rsv in HeLa S3 cells, and indicated that they are under the control of the duplicated GGAA (TTCC) motifs, which are contained in their 5’-upstream regions [11,14]. Rsv, which is a natural compound that is contained in red wine, grape skins, and peanuts, not only elongates the life span and health span of various species [43], but is also expected to be a lead compound with anti-cancer/tumor effects [44,45]. Rsv affects a number of biologically-significant proteins, including sirtuins [46], cAMP phosphodiesterase [47], mitochondrial complex I [48], DEPTOR [49], COX-1 and -2 [50], to extend the lifespan [43].
The most essential effect of the Rsv is that it activates SIRT1, which is an NAD+-dependent deacetylase [12]. The biological relevance of the SIRT1/3 has been reviewed and discussed in the context of the multiple effectors that regulate PGC-1a, FoxO and other functional proteins [51]. The excessive production of the NAD+ molecule by the Rsv-activated mitochondrial complex I will upregulate the NAD+/NADH ratio to lead to the modification of chromatin-associating proteins [52] and modulate the gene expression in immune system [53]. It has been shown that the transcription of the bidirectional promoter-driven BRCA1/NBR2 genes is regulated by the metabolic switch, depending on the NAD+/NADH ratio, which could be elevated by CR mimetic drugs [54]. It was shown that the C terminal-binding protein (CtBP) [55,56] has a central role in this regulation as a metabolic sensor. The accumulated NAD+ molecule will be consumed by poly(ADP-ribose) polymerase (PARP) to induce the poly(ADP-ribosyl)ation of nuclear proteins, which is essential for DNA-repair synthesis and the regulation of the telomere length [57,58]. The binding of Rsv to a human tRNA synthetase (TyrRS) leads to activation of PARP1 to induce a stress response [59]. We previously reviewed the biological relevance of the NAD+ molecule, especially in relation to its pivotal roles in metabolism and the protection of chromosomal DNAs [60,61]. Recently, it was reported that nuclear poly(ADP-ribose) could be utilized by NUDIX5 to supply ATP molecules, which are required for chromatin remodeling [62]. Thus, Rsv might directly or indirectly evoke the DNA-repair system, leading to the accumulation of NAD+ molecules or the activation of the GGAA-motif binding TFs that are sensitive to the NAD+/NADH ratio. NAD+ and its precursor nicotinamide have been reported to ameliorate metabolism or the mitochondrial functions [63-65]. Recently, it was revealed that NAD+ repletion improves the mitochondrial function to enhance the life span of adult mouse stem cells [66]. Conversely, decreased concentrations of NAD+ could cause aging or aging-related diseases [67]. These observations are consistent with a number of reports that suggest that Rsv is beneficial for cells inducing NAD+ level. This effect of the Rsv might be associated with the induction of mitohormesis [68,69].
It has been suggested that mitochondrial dysfunction leads to the development of tumors or cancerous cells [70,71]. In breast cancer cells, the knockdown of the subunit NDUFV1 leads to an aberration in complex I, which was shown to enhance aggressiveness or metastasis [72]. The upregulation of the cellular NAD+ level, which could be induced by Rsv, may contribute to mitochondrial integrity to suppress oncogenesis. It has been shown that Rsv upregulates mitochondrial biogenesis by the activation of Sirt1-induced PGC-1a [73]. Moreover, PGC-1a has been shown to drive NAD+ biosynthesis and thereby induce stress resistance [74]. However, Rsv might reduce the level of sirtuin(s), especially when it was used for a prolonged period or at high doses [14,75], suggesting that negative feedback system could regulate the excessive sirtuin activity.
It is worth noting again that the NAD+ molecule is the substrate of the PARP enzyme, which is required for the DNA damage response and the DNA repair system [60]. PARP1 activation may induce mitochondrial dysfunction [76]. Conversely, the inhibition of the PARP1 enzyme ameliorates the mitochondrial metabolism through the activation of SIRT1 [77]. We observed that the expression of the PARP gene was negatively regulated when poly(ADP-ribose) glycohydrolase (PARG) siRNAs were introduced into HeLa S3 cells [78]. As expected, the 5’-upstream regions of both the human PARP1 and PARG genes commonly contain the duplicated GGAA-motifs [37]. Additionally, PARP1 targets and poly(ADP-risosyl)ates transcription elongation factor NELF to release the paused RNA pol II dependent transcription [79], suggesting that PARP1 itself contributes to NAD+-sensitive transcription. These observations imply that the NAD+/NADH ratio might contribute to the fine-tuning of the transcription of genes that encode NAD+ metabolism-associated DNA repair factors. In other words, an NAD+-dependent transcription controlling system, which might be regulated by metabolic switch TFs, including CtBP1, CtBP2 [54-56] and PARP1, is required for cells to regulate the mitochondrial function in response to DNA-damage induced stress.
Besides the NAD+-dependent/-sensitive gene expression system, Rsv could affect other TFs. GC-boxes are found in the 5’-flanking regions of the human TERT and WRN genes [42,80], the promoter activities of which are augmented by treatment with 2-deoxy-D-glucose (2DG) or Rsv in HeLa S3 cells [10,81]. GC-boxes, which are mainly recognized by transcription factor Sp1 [82], are commonly contained in the promoter regions of genes that encode shelterins, which play a part in the maintenance of telomeres [83]. The human SIRT1 promoter, which contains GC-boxes, positively responds to PPARδ, suggesting that the mitochondrial β-oxidation pathway regulates the transcription of genes whose promoters possess GC-boxes [84]. We recently reported the co-operative function of the GC-boxes with a duplicated GGAA-motif in the human HELB promoter region [14]. The 263-bp region of the human eNOS promoter, containing binding elements for Sp1and ETS family protein ELF1, responds to Rsv [85]. The co-location of GC-boxes and c-ETS binding elements has been identified in in the 5’-flanking regions of the human VE-cadherin (CDH5) [86] and presenilin 1 (PS1) genes [87]. Furthermore, an interaction between Sp1 and ETS-1 activates the promoter activity of the murine guanylyl cyclase/natriuretic peptide receptor-A-encoding gene, Npr1 [88]. The adjacent locations of the ETS-binding motif and GC-boxes are found in a number of human gene regulatory regions [30], suggesting that these genes are ready to respond to Rsv or other mitohormesis-inducing compounds. The nuclear respiratory factor 1 (NRF1) is a DNA binding protein, the consensus binding sequence of which is 5’-YGCGCAYGCGCR-3’, binds to the transcriptionally regulatory regions of the mitochondrial function-associated genes in human glioblastoma cells [89]. Several of the NRF1-traget promoters, including the ATM, BAG1, COX15, FANCL, PRKDC, SDHB, SDHD, HARS and WRN genes, commonly contain the duplicated GGAA (TTCC) sequences [36,37]. In a recent study, a CAP-SELEX analysis indicated that TF-pairs are very frequently (95%) mediated/facilitated by DNA [90]. Moreover, it was indicated that the binding sequences for two varieties of TF pairs contain GGAA, and that the binding sites for FOXO1 and ETS family proteins can be closely positioned [90]. FOXO1, which is one of the forkhead box family proteins, regulates the growth of the vascular endothelium cells by reducing glycolysis and mitochondrial respiration [91]. Seventy-three MYC signature genes are downregulated by the forced expression of the gain of function mutation of FOXO1. Among them, GGAA duplications are harbored within 500-bp from the most upstream regions of at least 10 genes APEX1, HSPD1/HSPE1, NBN, NPM1, TP53, RFC2, RPL22, TK1, and TYMS, suggesting that FOXO1 might negatively modulate the transcription of duplicated GGAA-containing promoter driven genes. Thus, the interaction or co-operation between GGAA-binding proteins and other TFs, including Sp1 and FOXO1, might play an important part in the positive or negative response to Rsv. In addition to the ETS family, which includes at least 27 member proteins [92], NF-kB/c-Rel, IRFs, TEAD4, and STATs, have also been shown to bind to the GGAA(TTCC)-core motif containing sequences [32-35,90]. A recent study showed that Rsv enhances the immune function, including the production of IL2, by activating NF-kB in immunosuppressed mice [93]. Rsv induces the binding of EGR1 and EGR3 to the promoter region of the SMPD1 gene to induce its expression in human leukemia and cancer cells [94]. Moreover, Rsv stimulates cyclic AMP response element-dependent gene transcription [95], suggesting that CREB and ATF2 are the candidate target-TFs for Rsv-induced signals. The downregulation of the phosphorylation of the STAT3/5 proteins by Rsv was reported in human renal carcinoma cell lines [96], suggesting that Rsv negatively regulates STAT3/5-dependent gene expression. Rsv can also downregulate the expression of genes by blocking cytokine-induced NF-kB activation [97], or inhibit the transcription of Snail and thereby reduce its binding to promoter regions [98]. These lines of evidence suggest that Rsv has both positive and negative effects on transcription according to the combination of duplicated GGAA and other cis-elements, such as the GC-box, FOXO1, CREB, and NRF1-binding sequences, in the promoter regions.
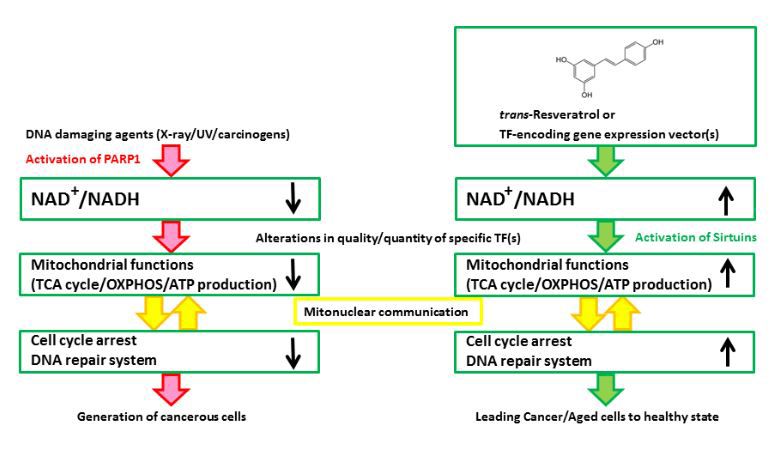
Figure 1. A schematic illustration of the hypothesized mechanism underlying the effects of Rsv or specific TF-encoding gene expression vector(s) on cancer/tumor cells. (Left) DNA damaging agents activate PARP1 to consume NAD+ for the synthesis of poly(ADP-ribose)s. Although PARP activity is required by the DNA repair system, the persistent reduction in the NAD+/NADH ratio will impair the progression of the TCA cycle, and the cells will produce ATP by glycolysis, which could be referred to as the “Warburg effect”. After the continuous activation of PARP1, cell cycle arrest and the DNA repair system no longer function well, and cells will proliferate regardless of the mitochondrial functions. (Right) Rsv will cause the cells to regain their NAD+/NADH level and activate sirtuins, NAD+ dependent transcription, and the mitochondrial functions. The cells will be altered, causing them to depend on the mitochondria, which control the regulation of energy production, cell cycle arrest, and DNA repair or telomeres in an appropriate manner. Rsv would thus directly ameliorate the mitochondria by upregulating the NAD+/NADH level, and indirectly induce the expression of the mitochondrial function/DNA repair-associated factor encoding genes. Hence, Rsv or some TF-encoding vectors are effective as tumor/aging suppressors.
It has been suggested that epigenetic alterations in chromosomes play essential roles in the generation and development of cancer cells [99-101]. In addition to NAD+, S-adenosylmethinine, acetyl-CoA, and other metabolites play important roles in DNA double-strand break (DSB) repair, by modulating chromosomal DNAs and proteins [102]. The aging processes are thought to be controlled by changes in DNA methylation and histone modifications in chromosomes [103-105]. These implications are consistent with the hypothesis that both oncogenesis and the aging process are controlled by both the accumulation of DNA damage [19, 106] and by epigenetic alterations [107, 108]. Thus, DNA methylation, histone modifications, chromatin organization/remodeling, and the regulation of coding/non-coding RNAs [109], should be taken into account when Rsv or novel anti-cancer drugs are administrated. Because Rsv is a potent activator of sirtuins, which are NAD+-dependent deacetylases or class III histone deacetylases (HDACs) [110], the alteration of the acetylation of histones and NF-kB [111, 112] could be induced by Rsv. A recent study suggested that Rsv enhances the effect of the FoxO1de-acetylation induced by fenofibrate (a SIRT1-upregulating agent), which upregulates the expression of SIRT1 [113]. A similar conclusion was obtained from an in vivo experiment, which showed a heart disease-protective effect of Rsv—in its suppression of proapoptotic signaling in the hearts of senescent mice [114]. To date, a number of HDAC inhibitors have been developed as anti-cancer or cancer preventing drugs [115, 116]. However, the regulation by Rsv might be indirect and maybe restricted to the target molecules that are de-acetylated by the sirtuins. The Rsv-induced signals partly affect the nutrient-associated signaling pathway [117], there by changes in the concentration of the acetyl-CoA molecule may affect the acetylation of nuclear proteins, including histone proteins. Thus, Rsv or sirtuin activators would be novel epigenetic controlling drugs that could have applications in cancer therapy.
The tumor hypoxia-induced inactivation of the TET enzyme, which catalyzes the demethylation of 5-methylcytosine (5mC), causes the hypermethylation of the promoter and enhancer regions of the tumor suppressor encoding genes [118]. Recently, genome-wide surveillance in breast cancer cells showed that both the hyper- and hypo-methylation of cancer-related genes occurred in response to Rsv treatment [119, 120]. In human liver cancer cells, Rsv induced the expression of the MAT2B gene, which encodes a methionine adenosyltransferase 2B [121]. Thus, the production of S-adenosylmethionine, which is widely known as methyl-group donor molecule, may have an impact on the methylation of chromosomal DNAs. The changes in the microRNA (miRNA) profile of white adipose tissue in response to Rsv treatment were investigated, and the upregulation of triacylglycerol metabolism-associated miRNAs was observed [122]. Among the miRNAs that were upregulated, miR-593-5p had the unique effect of downregulating the expression of SP1 protein, which suggests that Rsv could affect gene expression through the alteration of the cellular miRNA profile. The activities of telomerase and DNA methyltransferase (DNMT) are affected by Rsv treatment, implying that Rsv can induce genome maintenance signals in breast cancer cells [123]. We previously reported that telomerase activity is induced by Rsv in HeLa S3 cells [10]. The relationships between the telomeres and the risks of aging and aging-associated disease risks were reviewed and discussed [124]. Notably, telomere dysfunction compromises the mitochondria, causing them to overproduce reactive oxygen species (ROS) [125]. Thus, telomeres are associated with both the genome maintenance system and in communicating with the mitochondria [126]. Collectively, Rsv-induced signals may spontaneously link with the epigenetic alterations in chromosomes, to regulate the mitochondrial functions.
Various stresses, including oxidative stress and genotoxic stresses, are thought to be the main causes of cellular senescence [127]. Although it is widely believed that cancer is generated from the mutation of oncogenic driver genes, a number of observations, including cell cybrid experiments, strongly suggest that cancer originates from metabolic dysfunctions of the mitochondria [128]. In addition, it has been suggested that aging and neurodegeneration are impaired by mitochondrial deficiency when the dysfunction is mild, but that the effects of mitochondrial deficiency become deleterious when it is severe [129]. Recent studies have shown that Rsv prevents oxidative stresses in pancreatic β cells [130] and in neurons [131]. Rsv not only activates mitochondrial complex I [48] but also AMPK [132,133]. However, Rsv suppresses oxidative stress to prevent the dysfunction of endothelial cells [134]. It should be noted that the mitochondrial unfolded protein response (UPRmt), or a protective mitochondrial stress response, is induced by Rsv [135, 136]. The activation of the UPR may promote longevity and extend the life span [137]. In hematopoietic stem cells, with the help of NRF1, SIRT7 affects the UPRmt -mediated metabolic checkpoint to regulate the aging process, which is defined by the impairment of mitochondrial biogenesis, respiration, and cellular proliferation [138]. Of note, SIRT7 deacetylates GABPβ1, which is a master regulator of the nuclear-encoded mitochondrial function-associated genes [139].
Mitochondrial dysfunction is thought to be one of the reasons for oncogenesis [70, 71, 140]. A germline mutation analysis revealed that several of the TCA-cycle enzyme-encoding genes, including FH, SDHA to D, and SDHAF2, are tumor suppressor genes [141]. The essential role of complex I component NDUFV1 in the progression and metastasis of breast cancer has been shown [72]. Previously, FH (fumarate hydratase) and SDH (succinate dehydrogenase) were suggested to be tumor suppressors [142]. The knockdown of the citrate synthase encoding the Cs gene links the Warburg effect to tumor malignancy [143]. The downregulation of the mitochondrial complex 1 to 4- and Krebs cycle-associated gene expression in clear cell renal carcinoma (ccRCC) in comparison to normal kidney tissue was observed in a metabolic RNA-seq analysis [144]. Moreover, a decline in the mitochondrial quality or activity was associated with aging and the related diseases [41]. In this regard, the mitochondria represent the best organelle for directing next generation anti-cancer/aging therapy. Rsv upregulates the mitochondrial mass and ATP production in primary myoblasts [145]. The hypothesis that the mitochondria are the primary target of Rsv-induced signaling is also supported by the surveillance of the genomic sequences of the 5’-upstream regions of the human mitochondrial function-associated genes [36-38].
The results of a gene-wide survey analysis strongly suggest that the ancestor of the eukaryotic cells acquired the original a proteobacteria genome, which may be the origin of mitochondria, through endosymbiotic gene transfer (EGT) [146]. Alternatively, the acquisition of mitochondria, which carry energy production machinery, might have occurred after the incorporation of the genome of other ancestral bacteria [147]. Thus, the mitochondria have their own mtDNAs; however, almost all (99%) of the mitochondrial function-associated genes are present in the nuclear genome [148]. Remarkably, they are under the control of bi-directional promoters that have duplicated GGAA-motifs in common [36]. One possible reason why the mitochondrial function-associated genes are oriented head-head with other protein encoding genes might be that it was advantageous for the ancestral eukaryotes to incorporate bacterial DNAs into the nuclear genome using duplicated GGAA-motifs as landmarks. Actually, the purine-pyrimidine sequences, which are not only identified in the 5’-flanking regions of genes but also in sites involved in recombination, are known to affect the conformation of DNA helices [149]. Thus, duplicated GGAA-motifs might be the traces of the ancient transposable elements (TEs). This is a remarkable topic in the argument on how TFs help in the incorporation of the TEs [150]. The IFNG-responsive cis-regulatory elements, including the duplicated GGAA (TTCC) motif containing the GAS sequence, are thought to be remnants of retroviral enhancers [32]. Moreover, recent studies have shown that the binding sites of TFs are highly mutated in cancer cells [151,152]. Thus, an alternative explanation would be that ancestral eukaryotic cells might have acquired exogenous genes preferentially at the duplicated GGAA-motif-containing sites—where nuclear excision repair is impaired—through a long evolutionary process.
We have reported that the duplicated GGAA-motifs in the promoter regions of the human TP53 [11] and the HELB [14] genes play an essential role in the positive response to Rsv in HeLa S3 cells. The results are consistent with a previous study which showed the induction of growth arrest at the S-phase by Rsv in several human cancer cell lines [153]. Given that the duplicated GGAA-motif containing gene promoters could be upregulated by Rsv, the expression of the majority of the mitochondrial function-associated factor encoding genes may be synchronously regulated by Rsv-induced signaling in concert with the genes that encode DNA-repair-associated factors. Rsv may fulfill the stringent criteria for anticipated novel anti-cancer/aging drugs by inhibiting proliferation and improving the mitochondrial functions, to strengthen the DNA-repair system, with considerably fewer undesired side effects in comparison to the anti-cancer drugs that are in current use.
The execution of apoptosis is mediated by the mitochondria in response to various stresses including DNA-damage and immunological stress signals [154-156]. Thus, it has been proposed that the mitochondria serve as master regulators of danger signaling to determine cell death or survival [157]. A number of studies have shown that Rsv induces or regulates apoptosis [158]. Several mechanisms, including the regulation of the apoptosis regulators [159, 160] and miRNAs [161], are involved in the induction of apoptosis. On the contrary, Rsv has negative effect on apoptosis by suppressing the NF-kB signaling pathway [162]. The induction or suppression of apoptosis in response to Rsv might be dependent on the concentration or duration of treatment, or the inner cellular protein and RNA profiles. Surveillance of the human genomic DNA database indicated the presence of the duplicated GGAA-motifs in the 5’-regulatory regions of the human PDCD1, DFFA, BCL2, FAS, and FASL genes [27]. Moreover, the duplicated GGAA-motifs are contained in bidirectional promoters, such as the ATG12/AP3S1, APOPT1/BAG5, and HTRA2/AUP1 gene pairs [36]. Although it has been reported that ETS2 increases apoptosis in the brain and fibroblasts [163,164], the ETS family proteins, which bind to the GGAA (TTCC)-motif-containing sequences, are basically negative apoptosis regulators [39,165]. These observations suggest that the expression of the apoptosis related factor-encoding genes is modulated by the GGAA-duplicated sequences. Hence, genes that encode the executors and modulators of apoptosis are intrinsically upregulated by Rsv at the transcriptional level. However, occasional or temporal signals to induce apoptosis need to be avoided by certain signals that are regulated by the sirtuins, NF-kB and other proteins or miRNAs. Rsv might revoke both the signals that accelerate and decelerate apoptosis at the same time, ultimately bringing the cells to the point that they determine their own fate.
As previously described, the telomeres and mitochondria communicate with each other [126]. Several nuclear DNA-repair factors are suggested to play roles in the maintenance of mtDNAs, and damaged mtDNAs in turn exert signals to regulate nuclear transcription [166]. Recent in vivo experiments have shown that the mtDNA haplotype affects the mitochondrial morphology, functions, and aging parameters including the telomere length and UPRmt [167]. The mechanisms by which nuclear DNA damage signaling causes the mitochondrial dysfunctions that accelerate aging and aging-related diseases including cancer have been investigated in a review [168]. This process can be referred to as “mitonuclear communication” [169]. The nuclear DNA-repair systems need to work efficiently in order to maintain the mitochondrial function. Given that a proteobacteria are the putative ancestors of the mitochondria, the genome size must have been reduced by transferring genes into the ancestral eukaryotic cell nuclei. However, the mitochondria need to take care of the nuclear DNAs that contain almost all of their essential protein-encoding genes. Thus, the mitochondria might have developed a nuclear genome monitoring system, especially when DNA damaging signals are induced. The system by which DNA-repair/energy production is monitored might be mediated by the balance of the NAD+/NADH ratio, which is regulated by a number of enzymes in the nuclei, mitochondria, and cytosol [170]. In breast cancer cells, BRCA1 crosstalks with PARP1 to maintain a stable DNA repair ability, which would be partly sensitive to the NAD+ concentration [171]. The nucleus could be compared to a bank. We (the mitochondria) can conveniently deposit money (mitochondrial function-associated genes), but need to take care when a financial crisis (DNA damage) occurs. Rsv would represent an alert, that draws our interest toward the bank account.
In cancer cells, glycolysis is upregulated but the TCA cycle is downregulated [70,128]. Thus, inhibitors of glycolysis/PDHK1/PARP could be effective anti-cancer drugs targeting the metabolic switch in cancer cells [172]. It should be noted that Coenzyme Q (CoQ) can improve the mitochondrial functions [173]. Pdss2 KO mice, which carry complex I-III and II-III dysfunction due to insufficiency of CoQ or ubiquinone, show metabolic alterations and a Parkinson’s disease-like phenotype [174]. Defects in CoQ10 biosynthesis have been suggested to be one of the possible causes for glomerular or renal diseases [175]. We confirmed that the duplicated GGAA-motif, 5’-AAAGCTTCCGGGAGGAACTGGT-3’, is present near the TSS of the human PDSS2 gene, which encodes prenyl diphosphatase synthase subunit 2, implying that Rsv indirectly affects CoQ synthesis. These observations suggest that antioxidants, such as CoQ10 and vitamins C/E, would be lead drugs for anti-aging-associated disease, including Alzheimer’s disease [69,176]. Vitamin E or tocotrienols, and their analogues target mitochondria and the immune system to let cancer cells die [177]. Metformin and rapamycin are also expected to be novel anti-cancer/aging drugs that effectively suppress mTOR signaling [178]. A number of compounds that target mitochondria have been tested in clinical trials [179].
The introduction of gene expression vectors into cells or the editing of the genome of cells would represent an alternative approach to anti-cancer/aging therapy. As described above, Rsv can moderately upregulate the expression of various duplicated GGAA-motif driven genes, including the TP53 gene, suggesting that the expression vectors of the GGAA-motif binding proteins should be a primary target for examination. Moreover, given that the upregulation of the NAD+/NADH ratio improves the mitochondrial functions, the introduction of the redox reaction-associated genes could be applied in cancer treatment. The introduction of LbNOX oxidase (LbNOX) gene expression—which encodes an NADH oxidase—into HeLa cells via a lentiviral vector ameliorates the proliferative and metabolic defects caused by the impairment of the electron transport chain (ETC), which is associated with many human diseases [180]. For example, in the near future, skin grafting after the establishment of genome-edited iPS cells or direct gene transfer could be applied to the treatment of skin diseases, including melanoma [181]. The transfer of mitochondria or mtDNA into cells could be another therapeutic strategy for the treatment of mitochondrial dysfunction-related diseases [166,182,183]. If this therapy is applied on oocytes or fertilized eggs, the risk for cancer or aging processes could— in principle—be drastically reduced from birth. However, careful ethical consideration and safe and rigid therapeutic and technological guidelines are required for the handling of these cells.
In this article, we mainly focused on the relationships between the mitochondrial functions and the DNA repair systems, and suggested that they could both be controlled by Rsv at the transcriptional level. Although the molecular mechanisms underlying the regulation of the expression of these genes are not yet fully understood, several lines of evidence suggest that it is dependent on the NAD+/NADH balance or ratio.
Anti-cancer drugs that are in current use, including telomerase inhibitors, were developed with the intention of killing cancer cells. Although drugs immune receptor target drugs have been applied in the clinical setting, they are similar in that they attack cancer. Almost all of the anti-cancer drugs induce cell death not only in malignant cancer cells, but also in normal healthy cells. Sometimes the side effects are too severe to be ignored. In order to avoid lethal side effects, individual whole genome sequencing to find sensitivities to drugs, the development of a side effect monitoring system, and the improvement of treatment policies are required. These heavy tasks are necessitated by the intrinsic concept underlying the development and creation of most of the anti-cancer drugs that are in current use.
In the near future, a new concept for anti-cancer drugs or therapies must be developed and established. That is not to kill cancer cells but to force them to convert to a normal healthy state. Natural compounds, such as Rsv, or artificially generated compounds that ameliorate the mitochondrial functions and DNA repair systems to increase the NAD+/NADH level, are expected. Alternatively, specific TF expression vector(s) could be introduced into cancer cells to bring them back to the healthy state with both mitochondrial and chromosomal integrity. Somatic cells are able to gain pluripotency by the introduction of Yamanaka factors, all of which belong to DNA-binding TF. This implies that cancer/tumor cells could be forced to go back to the healthy state with the help of a TF(s). Designing anti-cancer/tumor drugs or gene transfer vector(s) based on this new concept will contribute to the prevention of aging and its associated diseases, including cancer.
We are grateful to Y. Ogawa, H. Hamada, H. Tabata, Y. Yamamoto, M. Kusaka, Y. Nakano and D. Sudo for outstanding technical assistance and discussion.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
- Chatterjee N, Siede W (2014) Replicating damaged DNA in eukaryotes. In: Friedberg EC, Elledge SJ, Lehmann AR, Lindahl T, Muzi-Falconi M, editors. DNA Repair, Mutagenesis, and Other Responses to DNA Damage. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, p. 331-349.
- Matson SW, Kaiser-Rogers KA (1990) DNA helicases. Annu Rev Biochem 59: 289-329. [Crossref]
- Browiec JA (1996) DNA helicases. In: DePamphilis ML, editor. DNA Replication in Eukaryotic Cells. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, p. 544-574.
- Menendez D, Inga A, Resnick MA (2009) The expanding universe of p53 targets. Nat Rev Cancer 9: 724-737. [Crossref]
- Kruiswijk F, Labuschagne CF, Vousden KH (2015) p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16: 393-405. [Crossref]
- Dyson NJ (2016) RB1: a prototype tumor suppressor and an enigma. Genes Dev 30: 1492-1502. [Crossref]
- Blackburn EH (2006) A history of telomere biology. In: de Lange T, Lundblad V, Blackburn E, editors. Telomeres (Second Edition). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, NY, p. 1-19.
- Vijg J (2007) Genome instability and accelerated aging. In: Vijg J, editor. Aging of the Genome. New York, NY: Oxford University Press, p. 151-180.
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, et al. (1996) Positional cloning of the Werner's syndrome gene. Science 272: 258-262. [Crossref]
- Uchiumi F, Watanabe T, Hasegawa S, Hoshi T, Higami Y, et al. (2011) The effect of resveratrol on the Werner syndrome RecQ helicase gene and telomerase activity. Curr Aging Sci 4: 1-7. [Crossref]
- Uchiumi F, Shoji K, Sasaki Y, Sasaki M, Sasaki Y, et al. (2015) Characterization of the 5’-flanking region of the human TP53 gene and its response to the natural compound, Resveratrol. J Biochem 159: 437-447. [Crossref]
- Wood JG, Rogina B, Lavul S, Howitz K, Helfand SL, et al. (2004) Sirtuin activators mimic caloric restriction and delay aging in metazoans. Nature 430: 686-689. [Crossref]
- Stefani M, Markus MA, Lin RC, Pinese M, Dawes IW, et al. (2007) The effect of resveratrol on a cell model of human aging. Ann NY Acad Sci 1114: 407-418. [Crossref]
- Uchiumi F, Arakawa J, Iwakoshi K, Ishibashi S, Tanuma S (2016) Characterization of the 5’-flanking region of the human DNA helicase B (HELB) gene and its response to trans-resveratrol. Sci Rep 6: 24510. [Crossref]
- Taneja P, Gu J, Peng R, Carrick R, Uchiumi F, et al. (2002) A dominant-negative mutant of human DNA helicase B blocks the onset of chromosomal DNA replication. J Biol Chem 277: 40853-40861. [Crossref]
- Gu J, Xia X, Yan P, Liu H, Podust VN, et al. (2004) Cell cycle-dependent regulation of a human DNA helicase that localizes in DNA damage foci. Mol Biol Cell 15: 3320-3332. [Crossref]
- Guler GD, Liu H, Vaithiyalingam S, Arnett DR, Kremmer E, et al. (2012) Human DNA helicase B (HDHB) binds to replication protein A and facilitates cellular recovery from replication stress. J Biol Chem 287: 6469-6481. [Crossref]
- TkÃ¡Ä J, Xu G2, Adhikary H3, Young JT, Gallo D4, et al. (2016) HELB Is a Feedback Inhibitor of DNA End Resection. Mol Cell 61: 405-418. [Crossref]
- Vijg J (2007) The aging genome. In: Aging of the Genome; The Dual Role of DNA in Life and Death. Oxford, UK: Oxford University Press, p. 181-232.
- Gordon DJ, Barbie DA, d’Andrea AD, Pellman D (2011) Chapter 2, Mechanisms of genetic instability. In: DeVita VT Jr, Lawrence TS, Rosenberg SA, editors. Cancer, Principles & Practice of Oncology; Primer of the Molecular Biology of Cancer Philadelphia, PA: Lippincott Williams & Wilkins, Wolters Kluwer Business, p. 26-48.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, et al. (2013) Cancer genome landscapes. Science 339: 1546-1558. [Crossref]
- Rahman N (2014) Realizing the promise of cancer predisposition genes. Nature 505: 302-308. [Crossref]
- Benz CC, Yau C (2008) Aging, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer 8: 875-879. [Crossref]
- Martincorena I, Campbell PJ (2015) Somatic mutation in cancer and normal cells. Science 349: 1483-1489.
- Carey MF, Peterson CL, Smale ST (2009) Transcription and preinitiation complex assembly in vitro. In: Transcriptional Regulation in Eukaryotes. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, p. 439-538.
- Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E (2007) Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389: 52-65. [Crossref]
- Uchiumi F, Miyazaki S, Tanuma S (2011) The possible functions of duplicated ets (GGAA) motifs located near transcription start sites of various human genes. Cell Mol Life Sci 68: 2039-2051. [Crossref]
- Oikawa T, Yamada T (2003) Molecular biology of the Ets family of transcription factors. Gene 303: 11-34. [Crossref]
- Hsu T, Trojanowska M, Watson DK (2004) Ets proteins in biological control and cancer. J Cell Biochem 91: 896-903. [Crossref]
- Hollenhorst PC, Shah AA, Hopkins C, Graves BJ (2007) Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev 21: 1882-1894. [Crossref]
- Larsen S, Kawamoto S, Tanuma S, et al. (2015) The hematopoietic regulator, ELF-, enhances the transcriptional response to Interferon-β of the OAS1 anti-viral gene. Sci Rep 5: 17497. [Crossref]
- Chuong EB, Elde NC2, Feschotte C2 (2016) Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351: 1083-1087. [Crossref]
- Platanias LC (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5: 375-386. [Crossref]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, et al. (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553-557. [Crossref]
- Uchiumi F, Larsen S, Masumi A, Tanuma S (2013) The putative implications of duplicated GGAA-motifs located in the human interferon regulated genes (ISGs). In: iConcept Ed. Genomics I-Humans, Animals and Plants. Hong Kong, iConcept Press Ltd., p. 87-105.
- Uchiumi F, Fujikawa M, Miyazaki S, Tanuma S (2013) Implication of bidirectional promoters containing duplicated GGAA motifs of mitochondrial function-associated genes. AIMS Mol Sci 1: 1-26.
- Uchiumi F, Larsen S, Tanuma S (2015) Chapter 5, Transcriptional regulation of the human genes that encode DNA repair- and mitochondrial function-associated proteins. In: Chen C, editor. DNA Repair. Rijeka, Croatia: InTech-Open Access Publisher Inc., p.129-167.
- Uchiumi F, Larsen S, Tanuma S (2016) Chapter , Possible roles of a duplicated GGAA motif as a driver cis-element for cancer-associated genes. In: iConcept, editor. Understand Cancer – Research and Treatment. Hong Kong: iConcept Press Ltd., p. 1-25.
- Yang ZF, Drumea K2, Mott S3, Wang J, Rosmarin AG4 (2014) GABP transcription factor (nuclear respiratory factor 2) is required for mitochondrial biogenesis. Mol Cell Biol 34: 3194-3201. [Crossref]
- Ostan R, Lanzarini C2,3, Pini E4, Scurti M5, et al. (2015) Inflammaging and cancer: a challenge for the Mediterranean diet. Nutrients 7: 2589-2621. [Crossref]
- Sun N, Youle RJ2, Finkel T3 (2016) The Mitochondrial Basis of Aging. Mol Cell 61: 654-666. [Crossref]
- Uchiumi F, Higami Y, Tanuma S (2010) Regulations of telomerase activity and WRN gene expression. In: Gagnon AN, editor. Telomerase: Composition, Functions and Clinical Implications. Hauppauge, NY: Nova Science Publishers, p. 95-103.
- Bhullar KS, Hubbard BP (2015) Lifespan and healthspan extension by resveratrol. Biochim Biophys Acta 1852: 1209-1218. [Crossref]
- Kundu JK, Surh YJ (2008) Cancer chemopreventive and therapeutic potential of resveratrol: mechanistic perspectives. Cancer Lett 269: 243-261. [Crossref]
- Harikumar KB, Aggarwal BB (2008) Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle 7: 1020-1035. [Crossref]
- Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5: 493-506. [Crossref]
- Park SJ, Ahmad F, Philp A, Baar K, Williams T, et al. (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148: 421-433. [Crossref]
- Desquiret-Dumas V, Gueguen N, Leman G, Baron S, et al. (2012) Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J Biol Chem 288: 36662-36675. [Crossref]
- Liu M, Wilk SA, Wang A, Zhou L, Wang RH, et al. (2010) Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem 285: 36387-36394. [Crossref]
- Bhat KP, Pezzuto JM (2002) Cancer chemopreventive activity of resveratrol. Ann N Y Acad Sci 957: 210-229. [Crossref]
- Sack MN, Finkel T (2012) Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol 4. [Crossref]
- Liu J, Kim J, Oberdoerffer P (2013) Metabolic modulation of chromatin: implications for DNA repair and genomic integrity. Front Genet 4: 182. [Crossref]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG (2013) Fueling immunity: insights into metabolism and lymphocyte function. Science 342: 1242454. [Crossref]
- Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K (2010) Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol 17: 1406-1413. [Crossref]
- Chinnadurai G (2002) CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell 9: 213-224. [Crossref]
- Chinnadurai G (2009) The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res 69: 731-734. [Crossref]
- Gibson BA, Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13: 411-424. [Crossref]
- d'Adda di Fagagna F, Hande MP, Tong WM, Lansdorp PM, Wang ZQ, et al. (1999) Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet 23: 76-80. [Crossref]
- Sajish M, Schimmel P2 (2015) A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol. Nature 519: 370-373. [Crossref]
- Maruta H, Okita N, Takasawa R, Uchiumi F, Hatano T, et al. (2007) The involvement of ATP produced via (ADP-Ribose)n in the maintenance of DNA replication apparatus during DNA repair. Biol Pharm Bull 30: 447-450. [Crossref]
- Tanuma SI, Sato A, Oyama T, Yoshimori A, Abe H, et al. (2016) New Insights into the Roles of NAD+-Poly(ADP-ribose) Metabolism and Poly(ADP-ribose) Glycohydrolase. Curr Protein Pept Sci . [Crossref]
- Wright RH, Lioutas A, Le Dily F, Soronellas D, Pohl A, et al. (2016) ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 352: 1221-1225. [Crossref]
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, et al. (2012) The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15: 838-847. [Crossref]
- Han X, Tai H, Wang X, Wang Z, Zhou J, et al. (2016) AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 15: 416-427. [Crossref]
- Yang Y, Sauve AA2 (2016) NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta 1864: 1787-1800. [Crossref]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, et al. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352: 1436-1443. [Crossref]
- Verdin E (2015) NAD+ in aging, metabolism, and neurodegeneration. Science 350: 1208-1213. [Crossref]
- Yun J, Finkel T (2014) Mitohormesis. Cell Metab 19: 757-766. [Crossref]
- Conti V, Izzo V, Corbi G, Russomanno G, Manzo V, et al. (2016) Antioxidant Supplementation in the Treatment of Aging-Associated Diseases. Front Pharmacol 7: 24. [Crossref]
- Seyfried TN, Flores RE, Poff AM, D'Agostino DP (2014) Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 35: 515-527. [Crossref]
- Vyas S, Zaganjor E, Haigis MC (2016) Mitochondria and Cancer. Cell 166: 555-566. [Crossref]
- Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, et al. (2013) Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest 123: 1068-1081. [Crossref]
- Menzies KJ, Singh K, Saleem A, Hood DA (2013) Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288: 6968-6979. [Crossref]
- Tran MT, Zsengeller ZK, et al. (2016) PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528-532. [Crossref]
- Kilic Eren M, Kilincli A, Eren Ö (2015) Resveratrol Induced Premature Senescence Is Associated with DNA Damage Mediated SIRT1 and SIRT2 Down-Regulation. PLoS One 10: e0124837. [Crossref]
2021 Copyright OAT. All rights reserv
- Baxter P, Chen Y, Xu Y, Swanson RA (2014) Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl Stroke Res 5: 136-144. [Crossref]
- Bai P, Cantó C, Oudart H, Brunyanszki A, Cen Y, Thomas C, et al. (2011) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13: 461-468. [Crossref]
- Uchiumi F, Watanabe T, Ohta R, Abe H, Tanuma S (2013) PARP1 gene expression is downregulated by knockdown of PARG gene. Oncol Rep 29: 1683-1688. [Crossref]
- Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, et al. (2016) Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353: 45-50.
- Uchiumi F, Watanabe T, Tanuma S (2010) Characterization of various promoter regions of the human DNA helicase-encoding genes and identification of duplicated ets (GGAA) motifs as an essential transcription regulatory element. Exp Cell Res 316: 1523-1534. [Crossref]
- Zhou B, Ikejima T, Watanabe T, Iwakoshi K, Idei Y, et al. (2009) The effect of 2-deoxy-D-glucose on Werner syndrome RecQ helicase gene. FEBS Lett 583: 1331-1336. [Crossref]
- Wierstra I (2008) Sp1: emerging roles--beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 372: 1-13. [Crossref]
- Uchiumi F, Oyama T, Ozaki K, Tanuma S (2011) Characterization of 5’-flanking regions of various human telomere maintenance factor-encoding genes. In: Kruman I, editor. DNA repair. Rijeka, Croatia: InTech-Open Access Publisher Inc., p. 585-596.
- Okazaki M, Iwasaki Y, Nishiyama M, Taguchi T, Tsugita M, et al. (2010) PPAR βδ regulates the human SIRT1 gene transcription via Sp1. Endocr J 57: 403-413. [Crossref]
- Wallerath T, Deckert G, Ternes T, Anderson H, Li H, et al. (2002) Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation 106: 1652-1658. [Crossref]
- Gory S, Dalmon J, Prandini MH, Kortulewski T, de Launoit Y, et al. (1998) Requirement of a GT box (Sp1 site) and two Ets binding sites for vascular endothelial cadherin gene transcription. J Biol Chem 273: 6750-6755. [Crossref]
- Pastorcic M, Das HK (2004) Alternative initiation of transcription of the human presenilin 1 gene in SH-SY5Y and SK-N-SH cells. The role of Ets factors in the regulation of presenilin 1. Eur J Biochem 271: 4485-4494. [Crossref]
- Kumar P, Garg R, Bolden G, Pandey KN (2010) Interactive roles of Ets-, Sp, and acetylated histones in the retinoic acid-dependent activation of guanylyl cyclase/atrial natriuretic peptide receptor-A gene transcription. J Biol Chem 285: 37521-37530. [Crossref]
- Satoh J, Kawana N, Yamamoto Y (2013) Pathway analysis of ChIP-seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul System Biol 7: 139-152. [Crossref]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A2, et al. (2015) DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527: 384-388. [Crossref]
- Wilhelm K, Happel K, Eelen G2,3, Schoors S2, et al. (2016) FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529: 216-220. [Crossref]
- Wei GH, Badis G, Berger MF, Kivioja T, Palin K, et al. (2010) Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29: 2147-2160. [Crossref]
- Lai X, Pei Q, Song X, Zhou X, Yin Z, et al. (2016) The enhancement of immune function and activation of NFκB by resveratrol-treatment in immunosuppressive mice. Int Immunopharmacol 33: 42-47. [Crossref]
- Mizutani N, Omori Y, Kawamoto Y, Sobue S, Ichihara M, et al. (2016) Resveratrol-induced transcriptional up-regulation of ASMase (SMPD1) of human leukemia and cancer cells. Biochem Biophys Res Commun 470: 851-856.
- Thiel G, Rössler OG (2016) Resveratrol stimulates cyclic AMP response element mediated gene transcription. Mol Nutr Food Res 60: 256-265. [Crossref]
- Kim C, Beak SH, Um JY, Shim BS, Ahn KS (2016) Resveratrol attenuates constitutive STAT3 and STAT5 activation through induction of PTP? and SHP-2 tyrosine phosphatases and potentiates sorafenib-induced apoptosis in renal cell carcinoma. BMC Nephrol 17: 19.
- Kutty RK, Samuel W, Abay R, Cherukuri A, Nagineni CN, et al. (2015) Resveratrol attenuates CXCL11 expression induced by proinflammatory cytokines in retinal pigment epithelial cells. Cytokine 74: 335-338. [Crossref]
- Ji Q, Liu X, Han Z, Zhou L, Sui H, et al. (2015) Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-ß1/Smads signaling pathway mediated Snail/E-cadherin expression. BMC Cancer 15: 97. [Crossref]
- Feinberg AP, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4: 143-153. [Crossref]
- Timp W, Feinberg AP (2013) Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 13: 497-510. [Crossref]
- Beekman R, Kulis M, Martín-Subero JI (2016) The DNA methylomes of cancer. In: Fraga M, Fernández AF, editors. Epigenomics in Health and Disease. London, UK: Academic Press, Elsevier Inc., p. 183-207.
- Liu J, Kim J, Oberdoerffer P (2013) Metabolic modulation of chromatin: implications for DNA repair and genomic integrity. Front Genet 4: 182. [Crossref]
- Johnson KC, Christensen BC (2016) Genome-wide DNA methylation changes during aging. In: Fraga M, Fernández AF, editors. Epigenomics in Health and Disease. London, UK: Academic Press, Elsevier Inc., p. 127-144.
- Bainor AJ, David G (2016) The dynamics of histone modifications during aging. In: Fraga M, Fernández AF, editors. Epigenomics in Health and Disease. London, UK: Academic Press, Elsevier Inc., p. 145-162.
- Benayoun BA, Pollina EA, Brunet A (2015) Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 16: 593-610. [Crossref]
- Serrano M, Blasco MA (2007) Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol 8: 715-722. [Crossref]
- Klutstein M1, Nejman D1, Greenfield R1, Cedar H (2016) DNA Methylation in Cancer and Aging. Cancer Res 76: 3446-3450. [Crossref]
- Sierra MI, Fernández AF, Fraga MF (2015) Epigenetics of Aging. Curr Genomics 16: 435-440. [Crossref]
- Blakey CA, Litt MD (2016) Epigenetic gene expression-an introduction. In: Huang S, Litt MD, Blakey CA, editors. Epigenetic Gene Expression and Regulation. London, UK: Academic Press, Elsevier Inc., p. 1-16.
- Wu X, Cao N, Fenech M, Wang X (2016) Role of Sirtuins in Maintenance of Genomic Stability: Relevance to Cancer and Healthy Aging. DNA Cell Biol 35: 542-575. [Crossref]
- Venturelli S, Berger A, Böcker A, Busch C, Weiland T, et al. (2013) Resveratrol as a Pan-HDAC inhibitor alters the acetylation status of histone proteins in human-derived hepatoblastoma cells. PLoS One 8: e73097. [Crossref]
- Bagul PK, Deepthi N, Sultana R, Banerjee SK (2015) Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J Nutr Biochem 26: 1298-1307. [Crossref]
- Wang WR, Liu EQ, Zhang JY, Li YX, Yang XF, et al. (2015) Activation of PPAR alpha by fenofibrate inhibits apoptosis in vascular adventitial fibroblasts partly through SIRT1-mediated deactylation of FoxO1. Exp Cell Res 338: 54-63. [Crossref]
- Sin TK, Yu AP, Yung BY, Yip SP, Chan LW, et al. (2014) Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signaling in senescent heart. J Physiol 592: 2535-2548.
- Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6: 38-51. [Crossref]
- Yan B, Li X, Johnson A, Yang Y, Jian W, et al. (2016) Epigenetic drugs for cancer therapy. In: Huang S, Litt MD, Blakey CA, editors. Epigenetic Gene Expression and Regulation. London, UK: Academic Press, Elsevier Inc., p. 397-423.
- Gasparrini M, Giampieri F, M Alvarez Suarez J, Mazzoni L, Y Forbes Hernandez T, et al. (2016) AMPK as a new attractive therapeutic target for disease prevention: the role of dietary compounds AMPK and disease prevention. Curr Drug Targets 17: 865-889. [Crossref]
- Thienpont B, Steinbacher J, Zhao H, D'Anna F, Kuchnio A, et al. (2016) Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537: 63-68. [Crossref]
- Medina-Aguilar R, Pérez-Plasencia C, Marchat LA, Gariglio P, García Mena J, et al. (2016) Methylation Landscape of Human Breast Cancer Cells in Response to Dietary Compound Resveratrol. PLoS One 11: e0157866. [Crossref]
- Lubecka K, Kurzava L, Flower K, Buvala H, Zhang H, et al. (2016) Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 37: 656-668. [Crossref]
- Yang H, Zheng Y, Li TWH, Peng H, Fernandez-Ramos D, et al. (2013) Methionine adenosyltransferase 2B, HuR, and Sirtuin 1 protein cross-talk impacts on the effect of resveratrol on apoptosis and growth in liver cancer cells. J Biol Chem 288: 23161-23170. [Crossref]
- Gracia A, Miranda J, Fernández-Quintela A, Eseberri I, Garcia-Lacarte M, et al. (2016) Involvement of miR-539-5p in the inhibition of de novo lipogenesis induced by resveratrol in white adipose tissue. Food Funct 7: 1680-1688. [Crossref]
- Kala R, Shah HN, Martin S, Tollefsbol TO (2015) Epigenetic-based combinatorial resveratrol and pterostilben alters DNA damage response by affecting SIRT1 and DNMT enzyme expression, including SIRT1-dependent ?-H2AX and telomerase regulation in triple-negative breast cancer. BMC Cancer 15: 672. [Crossref]
- Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 350: 1193-1198. [Crossref]
- Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, et al. (2011) Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470: 359-365. [Crossref]
- Sahin E, DePinho RA (2012) Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol 13: 397-404. [Crossref]
- Campisi J1, d'Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729-740. [Crossref]
- Seyfried TN (2015) Cancer as a mitochondrial metabolic disease. Front Cell Dev Biol 3: 43. [Crossref]
- Troulinaki K, Bano D (2012) Mitochondrial deficiency: a double-edged sword for aging and neurodegeneration. Front Genet 3: 244. [Crossref]
- Lee YE, Kim JW, Lee EM, Ahn YB, Song KH, et al. (2012) Chronic resveratrol treatment protects pancreatic islets against oxidative stress in db/db mice. PLoS One 7: e50412. [Crossref]
- Bernier M, Wahl D, Ali A, Allard J, Faulkner S, et al. (2016) Resveratrol supplementation confers neuroprotection in cortical brain tissue of nonhuman primates fed a high-fat/sucrose diet. Aging (Albany, NY) 8: 899-916.
- Yun H, Park S, Kim MJ, Yang WK, Im DU, et al. (2014) AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J 281: 4421-4438.
- Thompson AM, Martin KA, Rzucidlo EM (2014) Resveratrol induces vascular smooth muscle cell differentiation through stimulation of SirT1 and AMPK. PLoS One 9: e85495. [Crossref]
- Hu M, Liu B (2016) Resveratrol via activation of LKB1-AMPK signaling suppresses oxidative stress to prevent endothelial dysfunction in diabetic mice. Clin Exp Hypertens 38: 381-387. [Crossref]
- Mouchiroud L Houtkooper RH, Moullan N, Katsuyuba E, Ryu D, et al. (2013) The NAD+/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154: 430-441.
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, et al. (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497: 451-457.
- Wang Y, Hekimi S (2015) Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science 350: 1204-1207. [Crossref]
- Mohrin M, Shin J, Liu Y, Brown K, Luo H, et al. (2015) Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347: 1374-1377. [Crossref]
- Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, et al. (2014) A SIRT7-dependent acetylation switch of GABPß1 controls mitochondrial function. Cell Metab 20: 856-869. [Crossref
- Dong LF, Neuzil J (2014) Mitochondria in cancer: why mitochondria are a good target for cancer therapy. Prog Mol Biol Transl Sci 127: 211-227. [Crossref]
- Rahman N (2014) Realizing the promise of cancer predisposition genes. Nature 505: 302-308. [Crossref]
- Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, et al. (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14: 2231-2239. [Crossref]
- Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, et al. (2012) Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep 2: 785. [Crossref]
- Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, et al. (2014) Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513: 251-255. [Crossref]
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, et al. (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339: 1216-1219. [Crossref]
- Ku C, Nelson-Sathi S, Roettger M, Sousa FL, Lockhart PJ, et al. (2015) Endosymbiotic origin and differential loss of eukaryotic genes. Nature 524: 427-432. [Crossref]
- Pittis AA, Gabaldón T (2016) Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry. Nature 531: 101-104.
- Vafai SB, Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491: 374-383. [Crossref]
- Hanvey JC, Klysik J, Wells RD (1988) Influence of DNA sequence on the formation of non-B right-handed helices in oligopurine-oligopyrimidine inserts in plasmids. J Biol Chem 263: 7386-7396.
- Lynch VJ (2016) GENETICS. A copy-and-paste gene regulatory network. Science 351: 1029-1030. [Crossref]
- Perera D, Poulos RC, Shah A, Beck D, Pimanda JE, et al. (2016) Differential DNA repair underlies mutation hotspots at active promoters in cancer genomes. Nature 532: 259-263. [Crossref]
- Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A, López-Bigas N, et al. (2016) Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532: 264-267. [Crossref]
- Joe AK, Liu H, Suzui M, Vural ME, Xiao D, et al. (2002) Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin Cancer Res 8: 893-903. [Crossref]
- Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11: 621-632. [Crossref]
- Estaquier J, Vallette F, Vayssiere JL, Mignotte B (2012) The mitochondrial pathways of apoptosis. In: Scatena R, Bottoni P, Giardina B, editors. Advances in Mitochondrial Medicine, Dordrecht, Germany: Springer Science+Business Media BV, p. 157-183.
- Kroemer G (1997) Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ 4: 443-456. [Crossref]
- Galluzzi L1, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 13: 780-788. [Crossref]
- Varoni EM, Lo Faro AF, Sharifi-Rad J, Iriti M (2016) Anticancer Molecular Mechanisms of Resveratrol. Front Nutr 3: 8. [Crossref]
- Shanker S, Chen Q, Siddiqui I, Sarva K, Srivastava RK (2007) s. J Mol Signal 2: 7.
- Shanker S, Siddiqui I, Srivastava RK (2007) Molecular mechanisms of resveratrol (3,4,5-trihydroxy-trans-stilbene) and its interaction with TNF-related apoptosis inducing ligand (TRAIL) in androgen-insensitive prostate cancer cells. Mol Cell Biochem 304: 273-285. [Crossref]
- Venkatadri R, Muni T, Iyer AK, Yakisich JS, Azad N (2016) Role of apoptosis-related miRNAs in resveratrol-induced breast cancer cell death. Cell Death Dis 7: e2104. [Crossref]
- Busch F, Mobasheri A, Shayan P, Lueders C, Stahlmann R, et al. (2012) Resveratrol modulates interleukin-1β-induced phosphatidylinositol 3-kinase and nuclear factor kB signaling pathways in human tenocytes. J Biol Chem 287: 38050-38063. [Crossref]
- Baldus CD, Liyanarachchi S, Mrozek K, Auer H, Tanner SM, et al. (2004) Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: Amplification discloses overexpression of APP, ETS, and ERG genes. Proc Natl Acad Sci USA 101: 3915-3920. [Crossref]
- Ge Y, LaFiura KM, Dombkowski AA, Chen Q, Payton SG, et al. (2008) The role of the proto-oncogene ETS2 in acute megakaryocytic leukemia biology and therapy. Leukemia 22: 521-529. [Crossref]
- Kar A, Gutierrez-Hartmann A (2013) Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit Rev Biochem Mol Biol 48: 522-543. [Crossref]
- Kazak L, Reyes A, Holt IJ (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13: 659-671. [Crossref]
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, et al. (2016) Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535: 561-565. [Crossref]
- Fei Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, et al. (2016) Nuclear DNA damage signaling to mitochondria in aging. Nat Mol Cell Biol 17: 308-321. [Crossref]
- Quiros PM, Mottis A, Auwerx J (2016) Mitonuclear communication in homeostasis and stress. Nat Mol Cell Biol 17: 213-226. [Crossref]
- Cantó C, Menzies KJ, Auwerx J (2015) NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab 22: 31-53. [Crossref]
- Li D, Bi FF, Chen NN, Cao JM, Sun WP, et al. (2014) A novel crosstalk between BRCA1 and poly (ADP-ribose) polymerase 1 in breast cancer. Cell Cycle 13: 3442-3449. [Crossref]
- Schulze A, Harris AL (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491: 364-373. [Crossref]
- Wang Y, Oxer D, Hekimi S (2015) Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun 6: 6393. [Crossref]
- Peng M, Ostrovsky J, Kwon YJ, Polyak E, Licata J, et al. (2015) Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum Mol Genet 24: 4829-4847. [Crossref]
- Emma F, Montini G, Parikh SM, Salviati L (2016) Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 12: 267-280. [Crossref]
- Ajith TA, Padmajanair G (2015) Mitochondrial Pharmaceutics: A New Therapeutic Strategy to Ameliorate Oxidative Stress in Alzheimer's Disease. Curr Aging Sci 8: 235-240. [Crossref]
- Dong LF, Neuzil J (2014) Chapter 7, Vitamin E analogues as prototypic mitochondria-targeting anti-cancer agents. In: Neuzil J, Pervaiz S, Fulda S, editors. Mitochondria: The Anti-cancer Target for the Third Millennium, Dordrecht, Germany: Springer Science+Business Media BV, p. 151-181.
- Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, et al. (2012) Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging (Albany NY) 4: 952-965. [Crossref]
- Chen G, Pelicano H, Ogasawara MA, Wang F, Huang P (2014) Chapter 8, Targeting mitochondria of cancer cells: mechanisms and compounds. In: Neuzil J, Pervaiz S, Fulda S, editors. Mitochondria: The Anti-cancer Target for the Third Millenium. Dordrecht, Germany: Springer Science+Business Media BV, p. 183-210.
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, et al. (2016) Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352: 231-235. [Crossref]
- Sun BK, Siprashvili Z, Khavari PA (2014) Advances in skin grafting and treatment of cutaneous wounds. Science 346: 941-945. [Crossref]
- Hamilton G (2015) The hidden risks for 'three-person' babies. Nature 525: 444-446. [Crossref]
- Wallace DC (2016) Genetics: Mitochondrial DNA in evolution and disease. Nature 535: 498-500. [Crossref]