Surfactant proteins A and D (SP-A and SP-D) are involved in innate immunity against various pathogens. Under normal conditions SP-A and SP-D can bind to signal regulatory protein α (SIRPα), inhibit p38 mitogen-activated protein kinase (p38 MAPK) activation. Previous study demonstrated that SP-A and SP-D double knockout (KO) mice have increased levels of phosphorylated p38 MAPK (p-p38 MAPK) compared to wild-type mice. In this study we studied effects of p38 MAPK activity and SP-D on in vivo phagocytosis of C. neoformans by alveolar macrophages using genetic modified murine model. SP-A and SP-D double KO, and humanized SP-D transgenic (hTG SP-D), and wild-type C57BL/6 mice (8-12 weeks) were used. Mice were treated with or without p38 inhibitor prior to intratracheal injection with 1 × 106 CFU/mouse of C. neoformans, and then mice were sacrificed six hours post-infection. The phagocytosis of C. neoformans was determined using phagocytic index of alveolar macrophages and number of colony-forming units (CFU) in BAL fluid. Data are means + SE and p<0.05 by t-test was considered significant. The results showed that basal level of p38 MAPK phosphorylation was significantly higher in KO mice than in WT mice; but it decreased to normal level after treating with p38 inhibitor in KO mice. In treatment with p38 inhibitor KO mice significantly decreased its ability of in vivo phagocytosis (42.8 ± 5.2) compared to the sham group (65.5 ± 9.03) (p<0.05). The treated group with p38 inhibitor showed higher CFU counts (496 ± 53.5) in the BALF compared to the control (274 ± 54.7) (p<0.05). Furthermore, transgenic SP-D expression in the hTG SP-D mice decreased p-p38 level and showed enhanced in vivo phagocytic activity of C. neoformans when compared to KO mice (p<0.05). We concluded that both p38 MAPK activation and SP-D play roles in the in vivo phagocytosis of C. neoformans.
SP-A, SP-D, C. neoformans, p38 MAPK activation, alveolar macrophages
Cryptococcus neoformans is an environmentally widespread yeast-like fungal pathogen that is responsible for significant morbidity and mortality in immunocompromised hosts. It is a leading cause of death within AIDS patients worldwide [1]. Infections caused by C. neoformans are increasing at an alarming rate due to the increase of immunocompromised individuals (AIDS patients, organ transplant recipients, and cancer patients) [2]. C. neoformans infection starts in the lung and in the immunocompetent, alveolar macrophages will most likely be the first immune cells to encounter cryptococci but in immune compromised host it can disseminate to other organs of the body [3,4]. In humans, inhalation of C. neoformans fungi can lead to pulmonary infection. Cryptococcal meningoencephalitis, a life-threatening complication, can result from hematogenous dissemination of C. neoformans from the lung to the central nervous system and usually requires aggressive chemotherapeutic intervention [5]. C. neoformans possesses several virulence factors to bypass the innate immune system. The polysaccharide capsule is the most important C. neoformans virulence factor [6,7]. Other virulence factors include phospholipase, which functions through destabilization of host cell membranes, urease, which alters pH, and proteinases, which degrade host proteins [8-10].
In nature, C. neoformans fungi possess minimal capsules and are therefore easily aerosolized. Due to the smaller size of the fungal cells in this state, the fungi can be inhaled to the level of the alveoli in the lung. The initial step in host defense, therefore, is the interaction between acapsular C. neoformans and alveolar macrophages [3,11]. Phagocytosis of encapsulated and acapsular C. neoformans by alveolar macrophages and other phagocytes has been examined by several investigators [12-14]. Opsonization of C. neoformans is an essential step in the process of phagocytosis by alveolar macrophages. IgG and C3b act as opsonins and greatly facilitate phagocytosis. However, the concentration of these serum opsonins in the lung might be below the sufficient level for the effective phagocytosis of C. neoformans by alveolar macrophages. Other opsonins in the alveolar spaces might play a role in this initial step of innate immune response against C. neoformans. Two candidate proteins, that posses opsonic functions in phagocytosis of many alveolar micro-organisms, are surfactant protein A (SP-A) and surfactant protein D (SP-D) [15,16]. SP-A and SP-D are members of the C-type lectin (collectin) family. They are involved in innate immunity and host defense against various pathogens and have been shown to opsonize and enhance the clearance of a number of microorganisms and allergens .They also play roles in the regulation of inflammatory processes [17-20].
SP-A and SP-D proteins consist of four domains: a) N-terminus, b) triple-helical collagen-like domain, c) neck region, d) carbohydrate recognition domain [21]. The mechanisms of SP-A and SP-D function during fungal infections are still unclear. There are conflicting results reported about the SP-A and SP-D function in the pathogenesis of C. neoformans infection; and only a few studies had been performed to evaluate the role of surfactant proteins in response to C. neoformans infection in vivo [22]. Schelenz et al. have demonstrated that SP-D agglutinates acapsular C. neoformans in vitro which might immobilize the pathogen in the lungs. By this mechanism, the alveolar macrophages could be attracted to the pathogen and involved in the initial control of cryptococcal infection. It has been reported that SP-A can stimulate chemotaxis of alveolar macrophages as well [23]. Other studies have shown that SP-A does not play a significant role in the host defense against the in vivo infection of C. neoformans [5]. Recent studies have demonstrated that pre-opsonization with SP-D enhances the phagocytosis of the acapsular cap59Δ mutant strain by macrophages and leads to the increased survival of both wild-type (H99) and cap59Δ cells [24].
In previous studies It has been shown that in the normal lungs, SP-A and SP-D bind to signal regulatory protein α (SIRPα) by their globular heads, and inhibit p38 mitogen-activated protein kinases (p38 MAPK) activation thus preventing the inflammatory mediator responses [25,26]. In this study we used genetically modified SP-A and SP-D murine model to study the roles of p38 MAPK activation and SP-D expression on the C. neoformans phagocytosis by alveolar macrophages in vivo. We found that both p38 MAPK activation and SP-D expression are important for macrophage mediated C. neoformans clearance in the lung.
Mice and infection model
Experiments were conducted using ten 8-12 weeks old male and female C57BL/6 mice, twenty one SP-A/D KO, and ten humanized SP-D transgenic mice (hTG SP-D).
The original SP-A/D KO mice with a C57BL/6 background were kindly provided by Dr. Hawgood, University of California San Francisco. Mice used in this study were bred in the animal core facility at SUNY Upstate Medical University under pathogen-free conditions. To generate humanized SP-D transgenic mice, a 5.3-kb DNA fragment consisting of a 3.7-kb human SP-C promoter, 1.2-kb human SP-D cDNA, and a 0.4-kb SV40 small t-intron poly(A) sequence was microinjected into fertilized oocytes from WT C57BL/6 mice. Human SP-D positive transgenic mice (hSP-D+, mSP-D+/+) were bred with SP-D KO mice to eliminate mouse SP-D gene background for at least five generations as described previously (Figure 1) [27]. All animal experiments were conducted in accordance with the Institutional Animal Care and use committee guidelines of SUNY Upstate Medical University and the National Institutes of Health and ARRIVE guidelines on the use of laboratory animals.
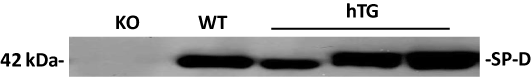
Figure 1. Human SP-D protein expression in the lung of humanized SP-D transgenic mice
SP-D expression in the BALF was examined by western blot analysis with hSP-D antibody. As expected, SP-A/D KO mouse did not express SP-D, transgenic mice express the level of SP-D comparable to that in WT mouse.
Mice were anesthetized by intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg). The mice were positioned on a taut string secured at one end, hanging from their incisors. C. neoformans fungi diluted in sterile saline to 1 × 106 yeast cells/50 μl were delivered into the lung by intratracheal injection. Anesthetized mice were monitored until full recovery. The mice were kept for six hours from the end of inoculation.
Mice were reanesthetized using intraperitoneal ketamine/xylazine (90 mg/kg ketamine, 10 mg/kg xylazine). After that they were euthanized by exsanguination. Then the midline neck incision was made on each mouse and the trachea was cannulated. The lungs were lavaged 2 times with 0.6 ml of sterile saline. 100 μl of bronchoalveolar lavage fluid (BALF) were taken for the determination of colony-forming units (CFUs). The rest of BAL fluid was centrifuged at 250xg for 10 min. The aliquot of cell-free BALF was stored at -20°C for further analysis. The pellet was resuspended in 200 μl of saline and centrifuged in the Hettich ROTOFIX 32A Benchtop centrifuge for 3 minutes to fix macrophages on a glass slide. The slides were left to dry for three hours then stained with hematoxylin and eosin (H&E) to examine the phagocytosis of C. neoformans [28].
Culture and maintenance of C. neoformans
C. neoformans serotype A wild-type strain H99 was used for this study. The cells from stock were streaked on yeast extract-peptone-dextrose (YPD) agarose plates and incubated at 37°C. Liquid cultures were grown in YPD medium at 37°C for 16 to 18 hours in a shaking incubator at 250 rpm. A single colony was taken from the plate, resuspended in saline and the number of yeast cells was determined by the hemocytometer. Yeast cells were prepared at 2 × 107 CFU/ ml and 50 μl (1 × 106 ) of yeast solution were used for each animal intratracheal injection.
Injection of p38 inhibitor
The p38 inhibitor (BIRB-796) (Sigma-Aldrich, St. Louis, MO) was prepared by adding 25 μl of the stock solution of the inhibitor (1 μg /μl) to 225 μl of normal saline. Then the total of 250 μl solution was injected intraperitoneally to each mouse one hour before the injection of the C. neoformans. The control group received 250 μl of normal saline in the same fashion.
BAL fluid
After obtaining the BALF, we take 100 μl for culture and the rest of BALF was centrifuged at 250xg for 10 minutes. The supernatant was kept in -20°C. Then, the pellet was reinstituted in 200 μl saline and centrifuged in the Hettich ROTOFIX 32A Benchtop centrifuge for 3 minutes and fix macrophages on glass slides.
Quantification of C. neoformans CFUs in the BALF
One hundred μl of BALF were aseptically cultured on YPD agar plates and the plates were then incubated at 37°C under aerobic conditions. After 48 hours of incubation, CFUs were assessed by using the Quantity One colony-counting software (Bio-Rad, Hercules, CA). Quantitative culture results were expressed as CFUs per milliliter of BAL.
Phagocytic analysis by Microscopy
The slides were stained for standard light microscopy using hematoxylin and eosin and were examined by light microscopy (Nikon Eclipse TE2000-U). The C. neoformans fungi were counted in 200 consecutive macrophages at x40 power fields and the total number of yeast cells was recorded for each mouse as phagocytic index.
Total protein concentration in homogenized lung tissue
The total protein concentration in homogenized lung tissue was measured using the micro BCA protein assay kit (Pierce Biochemicals, FL) as described previously [29]. Standard curves were prepared by using bovine serum albumin.
Western blot analysis for protein expression in lung
Lung tissues were homogenized in RIPA buffer which contains cocktail of protein inhibitors (Roche Molecular Biochemicals, IN). The lysates were centrifuged at 12,000 rpm for 15 min, and the supernatant was recovered for the Western blotting analysis as described previously [29]. In brief, the total proteins (8 μg/lane) from lung tissues of mice were subjected to gel electrophoresis (10% SDS-PAGE) under reducing conditions. The proteins in the gel were transferred onto polyvinylidene difluoride membrane. The membranes were probed with p38 antibody (Ab) (Santa Cruz Biotechnology, CA), p-p38 Ab (Santa Cruz Biotechnology, CA), and subsequently incubated with a secondary HRP-conjugated Ab (Bio-Rad, Hercules, CA). Bands were detected using ECL Western Blotting Detection Reagent (Pierce Biochemicals, FL) and the blots were exposed to X-film (Pierce Biochemicals, FL). Densitometry was carried out using ImageJ Software Version 1.48 (Wayne Rasband, National Institutes of Health, Bethesda, MD).
Statistical analysis
Data are expressed as means ± SE. Statistical analyses of the data were performed using SigmaStat software version 3.5 (Jandel Scientific, CA). Differences between/among groups were assessed by Student’s t test. p<0.05 was considered as statistically significant.
SP-A/D KO mice exhibit higher p-p38 MAPK activity in the lungs compared to WT mice
Both SP-A and SP-D proteins play important roles in the homeostasis and innate immunity in the lung. Under normal conditions SP-A and SP-D interact with several receptors on macrophage surface and inhibit p38 MAPK phosphorylation [30]. The data in this study showed that baseline p38 MAPK phosphorylation was significantly higher in the lung of SP-A/D KO compared to in WT mice (Figure 2A). After treatment with p38 inhibitor the level of p38 MAPK phosphorylation in the lung of SP-A/D KO mice was significantly reduced when compared to sham SP-A/D KO mice (Figure 2B). The graph in depicts the relative levels of p-p38 MAPK to unphosphorylated p38 MAPK in WT, hTG SP-D, and SP-A/D KO, with or without p38 inhibitor (Figure 2C).
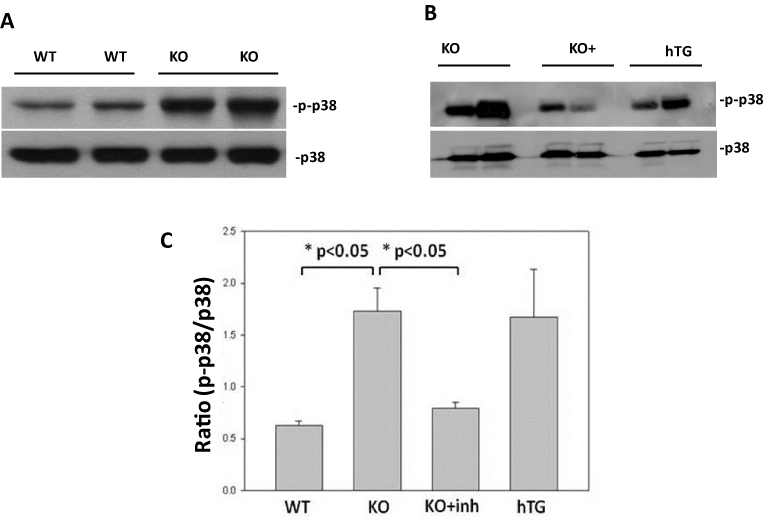
Figure 2. The levels of p38 MAPK phosphorylation in the lung of different types of mice
Total proteins of lung tissue were separated in 12% SDS-PAGE gel. Total p38 and phosphorated p38 (p-p38) were detected by western blotting analysis. The results showed: A. Higher level of phosphorylated p38 MAPK in KO mice compared to WT mice. B. Higher level of phosphorylated p38 MAPK in KO mice compared to p38 inhibitor-treated mice (KO inh = KO+ p38 inhibitor) and hTG SP-D mice (hTG). C. The graph depicts the relative levels of p-p38 MAPK to unphosphorylated p38 MAPK in WT, hTG SP-D, and SP-A/D KO, with or without p38 inhibitor.
Inhibition of p38 MAPK phosphorylation decreased in vivo phagocytosis of C. neoformans in SP-A/D KO mice
In order to examine the effect of p38 MAPK activation on phagocytosis of C. neoformans by alveolar macrophages, we first treated SP-A/D KO mice with p38 inhibitor (BirB 796) and then infected the mice intratracheally. The results showed that treated KO mice significantly reduced phagocytic activity of C. neoformans by macrophages in vivo (42.8 ± 5.2) compared to Sham group (65.5 ± 9.03) (p<0.05), suggesting that higher level of p-p38 activity in alveolar macrophages enhance in vivo phagocytosis of C. neoformans (Figure 3).
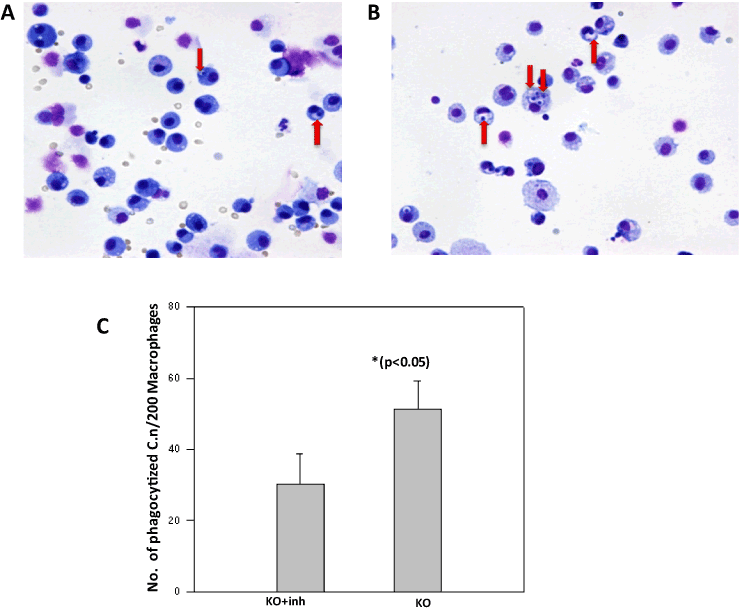
Figure 3. The effect of p38 MAPK activity on in vivo phagocytosis in KO mice
H&E stained macrophages showed the effect of p38 MAPK activity on in vivo phagocytosis of C. neoformans by alveolar macrophage. KO mice were treated with p38 inhibitor (A) or saline (sham) (B) one hour prior to yeast inoculation. Alveolar macrophages were prepared from the mice infected by C. neoformans (1 × 106 yeast cells/mouse) for 6 hours. The results demonstrated decreased phagocytic ability in the treated mice with p38 inhibitor compared to the sham KO mice. The graph (C) shows lower number of phagocytized yeast cells in 200 macrophages from p38 inhibitor-treated KO mice compared to in sham KO mice. Arrows point to engulfed C. neoformans.
Inhibition of p38 MAPK phosphorylation increased CFU counts C. neoformans in BAL fluid in SP-A/D KO mice
To examine whether there is difference of CFU counts of C. neoformans between treated and untreated SP-A/D KO mice we analyzed CFU number in the BALF from the mice infected 6 hrs. The results indicated that the p38 inhibitor treated group showed significantly higher CFU counts (496 ± 53.5) compared to the control (Sham) group (274 ± 54.7) (p<0.05) (Figure 4).
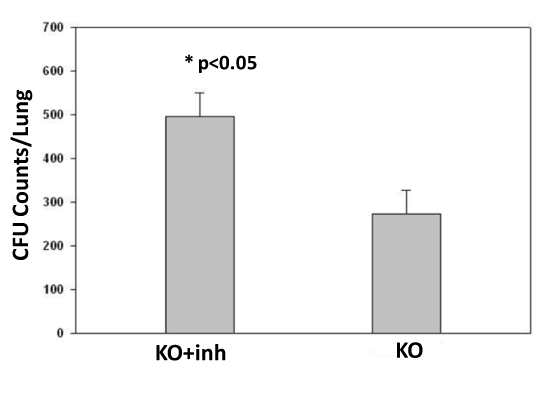
Figure 4. Comparison of CFU Counts in KO mice with or without p38 inhibitor treatment
BALFs were prepared from C. neoformans-infected KO mice with or without p38 inhibitor treatment. CFU counts were examined by culturing BALF on YPD agar media for 2 days. The p38 inhibitor-treated mice had greater CFU count than the sham mice (control) (p<0.05).
The phagocytic activity of C. neoformans by macrophages is significantly higher in hTG SP-D mice compared to KO mice
We have generated humanized SP-D transgenic mice which express human SP-D gene with mouse SP-A and SP-D KO background. To examine the role of SP-D in in vivo phagocytosis of C. neoformans in the present study we compared the phagocytic index as described in the method. We found that: A) The phagocytic index of hTG SP-D mice is higher than that of SP-A/D KO mice (101 ± 15.4 vs. 65.5 ± 9.03). B) The phagocytic index of hTG SP-D mice is higher than that of SP-A/D KO mice treated with p38 inhibitor (101 ± 15.4 vs. 42.8 ± 5.2) (p<0.05, Figure 5). These data demonstrated that both SP-D and p38 MAPK activation influence in vivo phagocytosis of C. neoformans by alveolar macrophages.
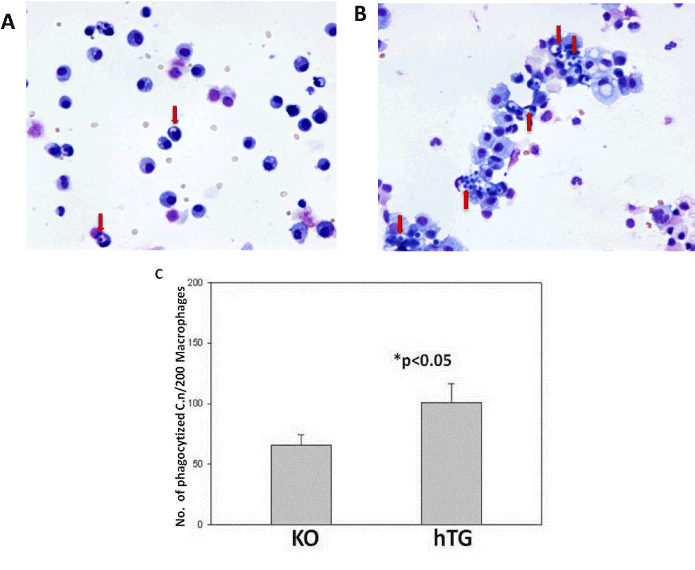
Figure 5. Effect of SP-D on in vivo phagocytosis in hTG and KO mice
H&E stained macrophages showed the effect of SP-D protein on in vivo phagocytosis of C. neoformans by alveolar macrophage. hTG SP-D and KO mice were infected intratracheally by C. neoformans (1 × 106 yeast cells/mouse) for 6 hours. Alveolar macrophages were prepared from C. neoformans-infected hTG SP-D and KO mice, respectively. Significantly greater numbers of engulfed C. neoformans were observed in hTG SP-D mice (B) compared to KO mice (A). Arrows point to engulfed yeasts in macrophages. The graph (C) shows significant difference in phagocytosis between the two groups (p<0.05).
Surfactant proteins SP-A and SP-D are important innate immune molecules in the lung and the roles have been extensively studied in pulmonary infection by various pathogens [31-34]. The role of SP-A and SP-D in fungal infection is controversial due to previous conflicting reports. It has been reported that in cases of Candida albicans infection, they bind to the pathogen and protect the lungs from infection [35]. SP-D has been shown to be protective in pulmonary infection and hypersensitivity reaction to Aspergillus fumigatus. [36-37]. However, the molecular and signaling mechanisms such p38 MAPK activation is not approached in previous works regarding C. neoformans infection and surfactant protein function [5,22-23,38]. In this work we first time found that the role of SP-D in the C. neoformans clearance through the regulation of p38 MAPK activation.
Van de Wetering et al., 2004 observed that SP-D had the higher affinity for acapsular C. neoformans and induced acapsular cryptococci aggregation. SP-D strongly bound encapsulated cryptococci through interaction with the cryptococcal capsular components glucuronoxylomannan (GXM) and mannoprotein 1 (MP1) [38]. Aggregation of lightly encapsulated C. neoformans by SP-D would be important in resistance at the initial stages of exposure to C. neoformans, especially in assisting clearance from the lung. It was shown that SP-D bound to the capsular components GXM and MPI and not to the other components. Increased capsular formation occurs shortly after infection, and more so during established infection. Shedding of GXM and its binding to SP-D could be a factor in host resistance. The capsule may thus be a way for Cryptococcus to escape innate immunity [39].
In the present work we found that SP-A/D KO mice have increased p-p38 MAPK level and showed higher phagocytic activity of alveolar macrophages for C. neoformans, suggesting the p-p38 MAPK signaling pathway may be involved in the processes of C. neoformans clearance in the lung infection. We further observed that the expression of transgenic SP-D gene significantly increase the ability of host clearance of C. neoformans, which is consistent with previous observation. The report by Geunes-Boyer et al. 2012 demonstrated that the presence of SP-D actually subvert the immune response against C. neoformans and that SP-A/D KO mice has a better survival rate after infection with C. neoformans [22]. In the early stages of infection, preopsonization with SP-D enhances phagocytosis of C. neoformans as it is clear in our study but preopsonization of the yeast cells with SP-D protected them against oxidative stress in both in vitro and in vivo situations. In another word, SP-D protects C. neoformans cells against host innate immune responses, particularly against the activity of oxidants.
SP-A and SP-D inhibit p38 MAPK phosphorylation and suppress inflammatory stimuli. In this study we found that inhibiting p38 MAPK activity in SP-A/D KO mice significantly decrease the phagocytic activity of alveolar macrophages and increase the fungal CFU count in BALF culture. These results are consistent with the findings of Gardai et al., 2003 that the surfactant Proteins A and D inhibit macrophage cytokine production and that SP-A and SP-D inhibit P38 activation [26]. In the normal lungs, SP-A and SP-D bind to signal regulatory protein α (SIRPα) of the macrophage by their CRD domains and inhibit p38 mitogen-activated protein kinases (p38 MAPK) activation. In the presence of pathogens, the CRD domain of SP-A and SP-D bind to the pathogen associated molecular patterns (PAMPs). The collagenous tails of SP-A and SP-D will interact with the macrophages’ calreticulin/CD91 to stimulate p38 phosphorylation and NF-κB activation, which leads to enhance phagocytosis of the pathogen [26].
Higher basal level of p38 MAPK phosphorylation in the SP-A/D KO mice may be due to the lack of this inhibitory mechanism as discussed above. The lack of SP-A and SP-D in the KO mice leads to higher activity of phosphorylated p38 which lead to increase in the phagocytic activity of the alveolar macrophages. Inhibition of p38 MAPK activation significantly decrease phagocytic activity of the alveolar macrophages in vivo.
In summary, we found that in the initial stages of C. neoformans infection, SP-D enhances phagocytosis by alveolar macrophages and that inhibition of p38 MAPK phosphorylation leads to significant decrease in the phagocytic activity of alveolar macrophages for C. neoformans in SP-A/D KO mice.
2021 Copyright OAT. All rights reserv
This study is supported by NIH HL096007.
- Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, et al. (2009) Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23: 525-530. [Crossref]
- Kennedy EN, Menon SK, West AH (2016) Extended N-terminal region of the essential phosphorelay signaling protein Ypd1 from Cryptococcus neoformans contributes to structural stability, phospho-stability, and binding of calcium ions. FEMS Yeast Res 16. [Crossref]
- Bojarczuk A, Miller KA, Hotham R, Lewis A, Ogryzko NV, et al. (2016) Cryptococcus neoformans Intracellular Proliferation and Capsule Size Determines Early Macrophage Control of Infection. Sci Rep 6: 21489. [Crossref]
- Perfect JR (1989) Cryptococcosis. Infect Dis Clin North Am 3: 77-102. [Crossref]
- Giles SS, Zaas AK, Reidy MF, Perfect JR, Wright JR (2007) Cryptococcus neoformans is resistant to surfactant protein A mediated host defense mechanisms. PLoS One 2: e1370. [Crossref]
- Dykstra MA, Friedman L, Murphy JW (1977) Capsule size of Cryptococcus neoformans: control and relationship to virulence. Infect Immun 16: 129-135. [Crossref]
- Gibson, JF, Johnston SA (2015) Immunity to Cryptococcus neoformans and C. gattii during cryptococcosis. Fungal Genet Biol 78: 76-86. [Crossref]
- Cox GM, Mukherjee J, Cole GT, Casadevall A, et al. (2000) Urease as a virulence factor in experimental cryptococcosis. Infect Immun 68: 443-448. [Crossref]
- Chen, LC, Blank ES, Casadevall A (1996) Extracellular proteinase activity of Cryptococcus neoformans. Clin Diagn Lab Immunol 3: 570-574. [Crossref]
- Pedroso RS, Lavrador MA, Ferreira JC, Candido RC, Maffei CM (2010) Cryptococcus neoformans var. grubii - Pathogenicity of environmental isolates correlated to virulence factors, susceptibility to fluconazole and molecular profile. Mem Inst Oswaldo Cruz 105: 993-1000. [Crossref]
- Mitchell TG, Perfect JR (1995) Cryptococcosis in the era of AIDS--100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev 8: 515-548. [Crossref]
- Bolanos B, Mitchell TG (1989) Phagocytosis of Cryptococcus neoformans by rat alveolar macrophages. Journal of medical and veterinary mycology 27: 203-217.
- Chaka W, Scharringa J, Verheul AF, Verhoef J, Van Strijp AG, et al. (1995) Quantitative analysis of phagocytosis and killing of Cryptococcus neoformans by human peripheral blood mononuclear cells by flow cytometry. Clin Diagn Lab Immunol 2: 753-759. [Crossref]
- Stukes S, Coelho C, Rivera J, Jedlicka AE, Hajjar KA, et al. (2016) The Membrane Phospholipid Binding Protein Annexin A2 Promotes Phagocytosis and Nonlytic Exocytosis of Cryptococcus neoformans and Impacts Survival in Fungal Infection. J Immunol 197: 1252-1261. [Crossref]
- Walenkamp AM, Verheul AF, Scharringa J, Hoepelman IM (1999) Pulmonary surfactant protein A binds to Cryptococcus neoformans without promoting phagocytosis. Eur J Clin Invest 29: 83-92. [Crossref]
- Wright JR (2005) Immunoregulatory functions of surfactant proteins. Nat Rev Immunol 5: 58-68. [Crossref]
- Erpenbeck VJ, Malherbe DC, Sommer S, Schmiedl A, Steinhilber W, et al. (2005) Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am J Physiol Lung Cell Mol Physiol 288: L692-698. [Crossref]
- Giannoni E, Sawa T, Allen L, Wiener-Kronish J, Hawgood S (2006) Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol 34: 704-710. [Crossref]
- LeVine AM, Elliott J, Whitsett JA, Srikiatkhachorn A, Crouch E, et al. (2004) Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am J Respir Cell Mol Biol 31: 193-199. [Crossref]
- Restrepo C, Dong Q, Savov J, Mariencheck WI, Wright JR (1999) Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am J Respir Cell Mol Biol 21: 576-585. [Crossref]
- Seaton BA, Crouch EC, McCormack FX, Head JF, Hartshorn KL, et al. (2010) Review: Structural determinants of pattern recognition by lung collectins. Innate Immun 16: 143-150. [Crossref]
- Geunes-Boyer S, Oliver TN, Janbon G, Lodge JK, Heitman J, et al. (2012) Surfactant protein D facilitates Cryptococcus neoformans infection. Infect Immun 80: 2444-2453. [Crossref]
- Schelenz S, Malhotra R, Sim RB, Holmskov U, Bancroft GJ (1995) Binding of host collectins to the pathogenic yeast Cryptococcus neoformans: human surfactant protein D acts as an agglutinin for acapsular yeast cells. Infect Immun 63: 3360-3366. [Crossref]
- Geunes-Boyer S, Oliver TN, Janbon G, Lodge JK, Heitman J, et al. (2009) Surfactant protein D increases phagocytosis of hypocapsular Cryptococcus neoformans by murine macrophages and enhances fungal survival. Infect Immun 77: 2783-2794. [Crossref]
- Waters P, Vaid M, Kishore U, Madan T (2009) Lung surfactant proteins A and D as pattern recognition proteins. Adv Exp Med Biol 653: 74-97. [Crossref]
- Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, et al. (2003) By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115: 13-23. [Crossref]
- Wang G, Guo X, Diangelo S, Thomas NJ, Floros J (2010) Humanized SFTPA1 and SFTPA2 transgenic mice reveal functional divergence of SP-A1 and SP-A2: formation of tubular myelin in vivo requires both gene products. J Biol Chem 285: 11998-2010. [Crossref]
- Xu Y, Ge L, Abdel-Razek O, Jain S, Liu Z, et al. (2016) Differential Susceptibility of Human Sp-B Genetic Variants on Lung Injury Caused by Bacterial Pneumonia and the Effect of a Chemically Modified Curcumin. Shock 45: 375-84. [Crossref]
- Liu J, Abdel-Razek O, Liu Z, Hu F, Zhou Q, et al. (2015) Role of surfactant proteins A and D in sepsis-induced acute kidney injury. Shock 43: 31-38. [Crossref]
- Hu F, Ding G, Zhang Z, Gatto LA, Hawgood S, et al. (2016) Innate immunity of surfactant proteins A and D in urinary tract infection with uropathogenic Escherichia coli. Innate Immun 22: 9-20. [Crossref]
- Bufler P, Schmidt B, Schikor D, Bauernfeind A, Crouch EC, et al. (2003) Surfactant protein A and D differently regulate the immune response to nonmucoid Pseudomonas aeruginosa and its lipopolysaccharide. Am J Respir Cell Mol Biol 28: 249-56. [Crossref]
- Gold JA, Hoshino Y, Tanaka N, Rom WN, Raju B, et al. (2004) Surfactant protein A modulates the inflammatory response in macrophages during tuberculosis. Infect Immun 72: 645-650. [Crossref]
- Hartshorn KL, Reid KB, White MR, Jensenius JC, Morris SM, et al. (1996) Neutrophil deactivation by influenza A viruses: mechanisms of protection after viral opsonization with collectins and hemagglutination-inhibiting antibodies. Blood 87: 3450-3461. [Crossref]
- van Iwaarden JF, van Strijp JA, Ebskamp MJ, Welmers AC, Verhoef J, et al. (1991) Surfactant protein A is opsonin in phagocytosis of herpes simplex virus type 1 by rat alveolar macrophages. Am J Physiol 261: L204-209. [Crossref]
- Rosseau S, Hammerl P, Maus U, Günther A, Seeger W, et al. (1999) Surfactant protein A down-regulates proinflammatory cytokine production evoked by Candida albicans in human alveolar macrophages and monocytes. J Immunol 163: 4495-4502. [Crossref]
- Madan T, Kishore U, Singh M, Strong P, Hussain EM, et al. (2001) Protective role of lung surfactant protein D in a murine model of invasive pulmonary aspergillosis. Infect Immun 69: 2728-2731. [Crossref]
- Madan T, Kishore U, Singh M, Strong P, Clark H, et al. (2001) Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest 107: 467-475. [Crossref]
- van de Wetering JK, Coenjaerts FE, Vaandrager AB, van Golde LM, Batenburg JJ (2004) Aggregation of Cryptococcus neoformans by surfactant protein D is inhibited by its capsular component glucuronoxylomannan. Infect Immun 72: 145-153. [Crossref]
- Brummer E, Stevens DA (2010) Collectins and fungal pathogens: roles of surfactant proteins and mannose binding lectin in host resistance. Med Mycol 48: 16-28. [Crossref]