seizures, neonate, pyridoxine dependent epilepsy, meningitis
Abbreviations
AED: Anti-epileptic Drug, CSF: Cerebrospinal Fluid, EC: emergency center, EEG: Electroencephalogram, NICU: Neonatal Intensive Care Unit, GBS: Group B Streptococcus, IV: Intravenous, MRI: Magnetic Resonance Imaging, MRA: Magnetic Resonance Angiography, MRV: Magnetic Resonance Venography, PDE: Pyridoxine-Dependent Epilepsy
A 4-day-old former 38-week term neonate was transferred to the NICU from a community emergency center (EC) due to concern for seizure activity. She was born at term via emergent Cesarean section for chorioamnionitis. The mother was group B streptococcus (GBS) positive and treated with clindamycin less than 4 hours before delivery. A blood culture was obtained which was negative. The baby did not receive postnatal antibiotics and was discharged home on the second day of life. On the third day of life, the mother witnessed an episode of whole body cyanosis with “stiffening of the arms and legs with jerking movements.” This prompted the parents to take the patient to the EC. At the community EC, the patient had respiratory distress with continued episodes of desaturations necessitating intubation. Rapid respiratory syncytial virus and influenza testing were negative. Patient also noted to exhibit upper and lower extremity flexion and extension concerning for seizure activity that transiently improved with Ativan 0.05 mg/kg. Blood cultures were obtained, and intravenous (IV) ampicillin and gentamicin were started. The patient was transferred to our level 4 NICU where electroencephalogram (EEG) showed multifocal sharp wave transients and depressed background activity in all regions. Antiepileptic treatment with Keppra 30 mg/kg/day divided three times a day was started. The blood cultures from the outside EC later grew Bacillus species in both aerobic and anaerobic culture bottles, but these had been considered a contaminant by the microbiology lab and were therefore discarded. Blood and CSF cultures obtained after treatment did not grow any pathogens, however there was a mild CSF leukocytosis (WBC 36). As no definitive bacteria were identified, the infant was treated for presumed meningitis with 14 days of IV vancomycin and cefotaxime. Magnetic resonance imaging (MRI) of the brain on day of life 4 showed multiple foci of restricted diffusion in the bilateral frontal lobes, right temporal lobe and bilateral pareito-occipital regions concerning for septic emboli or venous thrombosis (Figure 1A, B). A subsequent MRA/MRV was normal. Due to concern for hyper-coagulable state causing these MRI findings, hematology was consulted. After extensive testing, the results returned within normal limits. An echocardiogram also did not show any abnormalities. The patient’s seizure activity continued to worsen throughout the hospitalization despite increasing Keppra to 100 mg/kg/day and adding phenobarbital up to 8 mg/kg/day. The patient’s anti-epileptic drugs (AED) were ultimately escalated to include versed drip. EEG showed numerous right hemispheric electroclinical seizures (Figure 2). The patient had pyridoxine challenges on day of life 17 and day of life 28. Neither challenge produced an immediate change in clinical or electrographic seizures (Figure 3). After the second challenge, pyridoxine 50 mg/day was continued for 3 days with improvement in EEG (Figure 4). However, clinical improvement was believed to be related to changes in AEDs or improvement in meningitis rather than pyridoxine supplementation and pyridoxine was discontinued. Notably, repeat MRIs demonstrated improvement in the parietal-occipital lesions (Figure 1C, D) and repeat infectious work-ups including blood and CSF cultures remained negative. CSF neurotransmitters collected on hospital day 16 resulted 6 weeks later and showed an elevation of alpha-aminoadipic semialdehyde (AASA) suggesting the diagnosis of pyridoxine sensitive seizures. Pyridoxine (100 mg IV) was given and the patient was re-started on 100 mg (~30 mg/kg/day) of PO pyridoxine daily. The patient’s seizures improved significantly. Critical trio whole exome sequence with mitochondrial DNA resulted after discharge confirmed two pathogenic variants in ALDH7A1. The patient was maintained on pyridoxine supplementation and showed continued improvement in symptoms with concurrent ability to wean antiepileptic medication. She was discharged home following complete resolution of seizures with plan for further AED wean as an outpatient.
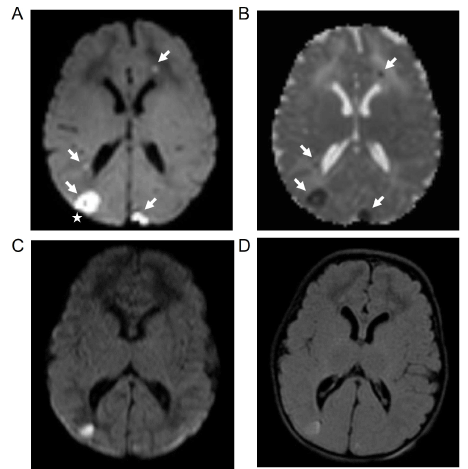
Figure 1. Axial diffusion weighted image MRI (A) and apparent diffusion coefficient MRI (B) on day of life 4 show multiple foci of restricted diffusion (arrows) with associated hemorrhage in the parieto-occipital foci (star). Axial diffusion weight image MRI (C) and FLAIR MRI (D) on day of life 16 show interval improvement of diffusion restriction.
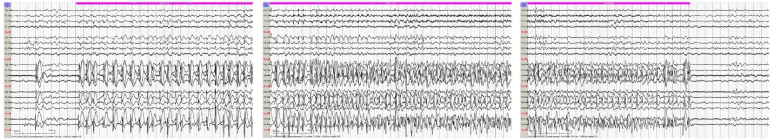
Figure 2. Sample EEG tracing showing electroclinical seizure starting in the right hemisphere captured on day of life 24.
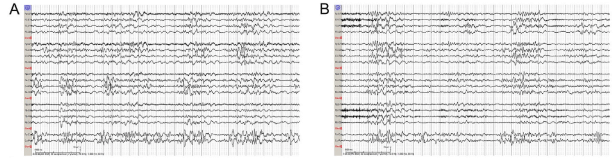
Figure 3. Sample EEG tracing before (A) and after (B) administration of 100 mg of IV pyridoxine shows no change in abnormal background pattern.
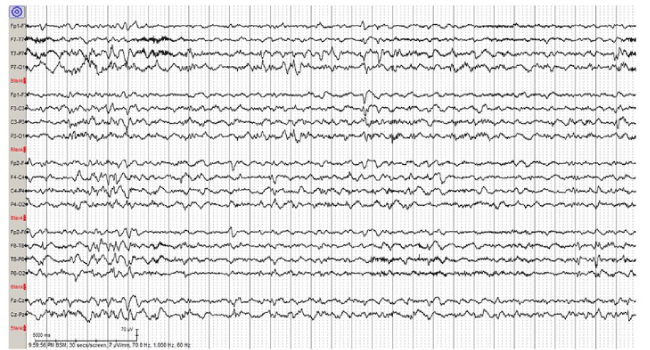
Figure 4. Sample EEG tracing from day of life 31 after initiation of PO pyridoxine shows improved background activity and no seizures.
Pyridoxine-dependent epilepsy (PDE) is a rare autosomal recessive condition that causes neonatal seizures with an incidence of 1 in 730,000 patients [1,2]. The condition occurs due to mutations in the gene encoding alpha-aminoadipic semialdehyde dehydrogenase (AASADH). When this enzyme is dysfunctional, alpha-aminoadipid semialdehyde accumulates and sequesters pyridoxine (vitamin B6). Therefore, supplementation with pyridoxine is imperative in the treatment of PDE to counter this depletion. Seizures typically occur soon after birth, but can also present up to 14 months of age or even as intrauterine convulsions [3-6]. Untreated, there is a high associated mortality rate [7]. Various seizure types have been associated with PDE ranging from focal myoclonic to generalized tonic-clonic seizures. EEG changes are also varied ranging from focal discharges to burst-suppression patterns, and confirmed electrographic seizures are not always present even though the neonate may have paroxysmal movements [3,8,9].
Suspicion for PDE should occur in several situations: in any child under the age of one year who has epilepsy and does not have any apparent brain malformations, in a previously normal neonate without abnormal perinatal history, in neonates with difficult-to-control seizures, or in infants with seizures that are only partially responsive to antiepileptic medications [10]. There may be a family history of epilepsy in siblings and/or parental consanguinity.
Evaluation for PDE is via either urine, plasma, or CSF evaluation of alpha-AASA or pipecolic acid elevations [10,11]. Once a test shows elevation of either or both of these markers, further genetic investigation evaluating for mutations in the ALDH7A1 gene on chromosome 5q31 can confirm the diagnosis. Diagnosis can also be made prenatally for fetuses suspected of having this disorder [4].
A clinical diagnosis can usually be made in a patient with seizures who receive 100 mg of IV pyridoxine while continuously monitoring the EEG and vital signs, avoiding the delay of genetic test results. In patients with PDE, the seizures typically resolve in just a few minutes after the pyridoxine supplementation although electrographic improvement may not be seen for several hours. If the EEG does not improve then a subsequent dose of up to 500 mg may be administered or oral pyridoxine can be empirically started. However, it is important to note that some patients have cardiovascular or neurologic depression following the pyridoxine supplementation. Therefore, it is imperative to monitor vital signs closely during this process [10]. Also of note, while the clinical seizures typically resolve immediately, in some cases the improvement may take longer. Regardless, clinical seizures should improve within one week of pyridoxine supplementation.
Neural imaging is commonly performed in patients with PDE, and brain malformations are increasingly being identified in these patients. Two main categories of brain anomalies are described: anatomic lesions versus lesions secondary to seizure activity. Anatomic lesions seen on MRI vary and include partial agenesis, hypoplasia or dysplasia of the corpus callosum, white matter anomalies, ventricular dilation or posterior fossa anomalies such as cerebellar hypoplasia [3,12,13]. The most common category is corpus callosum anomalies, affecting up to 42% of patients in one study [12]. In three studies with a total of 44 patients with PDE, normal MRIs were seen in 20-31%. There have been two postmortem neuropathological studies of confirmed PDE [14,15]. Jansen et al. reported severe dyslamination of the cortex with microcolumnar focal cortical dysplasia in resected tissue following right occipital and posterior parietal lobectomy at 15 months. The patient expired at 9 years old. Postmortem histological examination of remaining tissue revealed patchy cortical gliosis and increased amounts of interstitial neurons in
the white matter as well as hippocampic sclerosis which is known to occur with status epilepticus. Marguet et al. found similar findings in a child who died at 14 months of age [15]. Autopsy findings demonstrated dysgenesis of the corpus callosum, asymmetric ventricular dilatation, diffuse cortical vacuolization, neuronal loss, reactive gliosis, and areas of ischemic necrosis. Similar to the other report, hippocampic sclerosis was also found in this patient.
Treatment for PDE is pyridoxine supplementation which leads to control of both clinical and electrical seizure activity. However, even in patients that receive treatment, clinical outcomes can still be poor. Seventy-five percent of patients with PDE who received treatment had developmental delay or intellectual disability as defined by an intelligence quotient of less than 70 [12,16]. It is believed that long-term prognosis depends upon timing of treatment initiation. Studies on patients treated earlier suggest that they have higher functioning compared to those with later treatment [12]. A newer approach involves combining supplementation with pyridoxine, a lysine-restricted diet, and L-arginine supplementation [16-18].
In the setting of outside positive blood cultures, leukocytosis, and inadequately treated GBS positive mother, our term neonate’s clinical seizures were most concerning for meningitis. Although multiple infectious evaluations in our NICU were negative, the patient had already been treated with antibiotics. The patient’s continued seizure activity despite escalating AED in the setting of radiographic improvement on MRI prompted re-evaluation of other genetic forms of epilepsy. In retrospect, it is possible that the radiographic changes could be attributed to PDE itself. PDE has not been previously described in a neonate who was initially diagnosed with meningitis but later determined to have PDE. Of note, PDE was initially considered but the lack of immediate improvement on EEG focused the differential on other causes of epilepsy. This case report reinforces the need for evaluation of treatable forms of epilepsy such as PDE in a patient that exhibits continued and/or worsening seizures despite appropriate treatment even if an initial pyridoxine challenge has been negative. Unfortunately, there are no pathognomonic features of seizures or brain malformations that specifically point toward a diagnosis of PDE. Thus, a heightened suspicion for PDE must exist in seizing neonates in order to promptly and appropriately evaluate for it even if meningitis has already been diagnosed.
Funding source: No external funding for this manuscript.
Financial disclosure: None of the authors have financial relationships relevant to this article to disclose.
Conflict of interest: The authors have no potential conflicts of interest to disclose.
Clinical trial registration: None
Dr. Valentine helped conceptualize the manuscript, performed the literature search, drafted the manuscript, incorporated comments and approved the final manuscript for submission
Dr. Lyons-Warren and Dr. Seto each revised the drafted manuscript with an emphasis on the neurological perspective, prepared the figures and tables and approved the final manuscript for submission.
Dr. Davies helped conceptualize the manuscript, revised the drafted manuscript, and approved the final manuscript for submission.
- Baxter P, Griffiths P, Kelly T, Gardner-Medwin D (1996) Pyridoxine-dependent seizures: demographic, clinical, MRI and psychometric features, and effect of dose on intelligence quotient. Dev Med Child Neurol 38: 998-1006. [Crossref]
2021 Copyright OAT. All rights reserv
- Been JV, Bok LA, Andriessen P, Renier WO (2005) Epidemiology of pyridoxine dependent seizures in the Netherlands. Arch Dis Child 90: 1293-1296. [Crossref]
- Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, et al. (2010) Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain 133: 2148-2159. [Crossref]
- Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, et al. (2006) Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med 12: 307-309. [Crossref]
- Bejsovec M, Kulenda Z, Ponca E (1967) Familial intrauterine convulsions in pyridoxine dependency. Arch Dis Child 42: 201-207. [Crossref]
- van Karnebeek CD, Tiebout SA, Niermeijer J, Poll-The BT, Ghani A, et al. (2016) Pyridoxine-Dependent Epilepsy: An Expanding Clinical Spectrum. Pediatr Neurol 59: 6-12. [Crossref]
- Striano P, Battaglia S, Giordano L, Capovilla G, Beccaria F, et al. (2009) Two novel ALDH7A1 (antiquitin) splicing mutations associated with pyridoxine-dependent seizures. Epilepsia 50: 933-936. [Crossref]
- Hellstrom-Westas L, Blennow G, Rosen I (2002) Amplitude-integrated encephalography in pyridoxine-dependent seizures and pyridoxine-responsive seizures. Acta Paediatr 91: 977-980.
- Schmitt B, Baumgartner M, Mills PB, Clayton PT, Jakobs C, et al. (2010) Seizures and paroxysmal events: symptoms pointing to the diagnosis of pyridoxine-dependent epilepsy and pyridoxine phosphate oxidase deficiency. Dev Med Child Neurol 52: 133-142. [Crossref]
- Gospe SM, Pagon RA (1993) Pyridoxine-Dependent Epilepsy, in GeneReviews (R) Seattle (WA). [Crossref]
- Stockler S, Plecko B, Gospe SM Jr, Coulter-Mackie M, Connolly M, et al. (2011) Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab 104: 48-60. [Crossref]
- Bok LA, Halbertsma FJ, Houterman S, Wevers RA, Vreeswijk C, et al. (2012) Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol 54: 849-854. [Crossref]
- Perez B, et al., Clinical, biochemical, and molecular studies in pyridoxine-dependent epilepsy. Antisense therapy as possible new therapeutic option. Epilepsia 2013. 54: 239-248. [Crossref]
- Jansen LA, Hevner RF, Roden WH, Hahn SH, Jung S, et al. (2014) Glial localization of antiquitin: implications for pyridoxine-dependent epilepsy. Ann Neurol 75: 22-32. [Crossref]
- Marguet F, Barakizou H, Tebani A, Abily-Donval L, Torre S, et al. (2016) Pyridoxine-dependent epilepsy: report on three families with neuropathology. Metab Brain Dis 31: 1435-1443. [Crossref]
- van Karnebeek, CD, Jaggumantri S (2015) Current treatment and management of pyridoxine-dependent epilepsy. Curr Treat Options Neurol 17: 335. [Crossref]
- Coughlin CR 2nd, van Karnebeek CD, Al-Hertani W, Shuen AY, Jaggumantri S, et al. (2015) Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: Neurodevelopmental outcome. Mol Genet Metab 116: 35-43. [Crossref]
- van Karnebeek CD, Stockler-Ipsiroglu S, Jaggumantri S, Assmann B, Baxter P, et al. (2014) Lysine-Restricted Diet as Adjunct Therapy for Pyridoxine-Dependent Epilepsy: The PDE Consortium Consensus Recommendations. JIMD Rep 15: 1-11. [Crossref]