RIG-I (retinoic acid inducible gene-I) was ever identified as a cytoplasmic dsRNA [1] and uncapped 5′-triphosphate- ssRNA [2] virus sensor and activates downstream signaling, resulting in the induction of members of the type I interferon (IFN 1) family, which are regarded among the most important effectors of the innate immune system [3]. The notion generally leads some novices to take it for granted that RIG-I over-expression interferes with cell division and arrests cell cycle for viruses clearance, otherwise will result in infected cell necrosis, further acute inflammation. However, no related report on cell morphological change has seen us to date.
Molecular genetic analysis indicates that theoretically rig-i (9p12) [4] can cis-regulate the expression and secretion of type I IFN [ifn α1β (9p22), ifn β1(9p21)], and trans-regulate that of type II IFN [ifn γ (12q14)], further these cytokines can interfere with cell division and delay cell cycle. Thus it was presumed that virus infection stimulated the expression and secretion of RIG-I, IFNs and other inflammatory cytokines, usually further lead to inflammation [5]. In fact, previous substantial evidences indicated that this presumption is correct [1,6]. Inflammation is a crucial function of the innate immune system that protects against pathogens and initiates specific immunity. Acute inflammation is a rapid and self-limiting process: chemical mediators are induced in a tightly regulated sequence, and immune cells move in and out of the affected area, destroying infectious agents, repairing damaged tissue, and initiating a specific and long-term response to the pathogen. However, acute inflammation does not always resolve every threat to human body. Nine of ten diseases may be driven, at least in part, by chronic inflammation, and often subclinical inflammation [7]. For example, chronic inflammation is implicated in all stages of carcinogenesis, i.e., initiation, promotion and progression by several mechanisms including acceleration of cell cycle progression and cell proliferation, evasion from apoptotic cell death, and stimulation of tumor neovascularization [8]. Actually, these links have been confirmed in a number of murine models, especially in terms of gastric (H. pylori infection) [9], liver (cholangitis) [10] and colon (colitis) [11] cancers. In these and other animal cancer models [12], the cells and mediators of chronic inflammation act as tumor promoters at distinct phases of malignant progression. In brief, chronic inflammation increases risk of carcinogenesis, and many cancers arise at sites of chronic inflammation [13].
Growing evidence indicates that some inflammatory cytokines, e.g. IL-6, TNFα, etc. are the major molecular players involved in the inflammation-to-cancer axis [14]. What’s more, NF-kB has recently been identified as a potential molecular bridge between inflammation and cancer [11]. The induction of proinflammatory cytokines (e.g., IL-6 and TNF-a) is mediated via transcriptional activation of NF-kB. The NF-kB dependent activation of cell adhesion molecules, such as vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule (ICAM), which have been found to increase in various cancers, are involved in leukocyte adhesion and migration within the inflammatory tumor microenvironment. While the cytokine expression is regulated primarily by NF-kB, the tumor cell-derived cytokines further stimulate NF-kB-mediated transcription of proinflammatory genes in tumor cells, tumor-associated stromal cells and host tissues, thereby creating a sustained chronic inflammatory state within the tumor microenvironment. Actually, the role of NF-kB in chronic inflammation-driven tumor promotion has been shown in different experimental models. One example is that virus infection can up-regulate the expression of RIG-I in some cancers, further stimulate the expression and secretion of IFNs [19,20]. Our recent experiments indicated RIG-I over-expression elevated the expression of IL-6, TNFα and decreased the expression and activity of c-Myc (unpublished data). IL-6 mainly modulates the expression of genes involved in cell cycle progression and inhibition of apoptosis, primarily via JAK-STAT signaling pathway and Akt-mediated survival signaling pathway. An elevated level of IL-6 has been in the pathogenesis of various cancers. However, TNFα plays a dual role in carcinogenesis: high concentration always does destructive damage to tumor vasculature and causes necrosis, further results in inflammation [15]; while low concentration works as endogenous tumor promoter, and the expression of TNFα has been detected in various human cancers including breast, prostate, colon rectum, bladder, lymphona and leukemia. In addition, there were many evidences indicated that RIG-I may be an anti-oncogene, even though without direct proof till now.
A report confirmed that interaction between CARDs of RIG-I and IPS-1 results in the activation of type I IFNs and other proinflammatory cytokines [16], and phosphorylation of serine 8 located within the first CARD of RIG-I inhibits its ability to interact with TRIM25, and thus blunts IFN-β production (IPS-1-dependent RIG-I signal transduction). At present, IFNα2β, a member of the type I IFN family, is widely used as an anticancer drug against melanoma, however, its efficacy is significant but limited. Another research reported that a small RNA silenced the anti-apoptotic gene BCL-2 and also activated RIG-I. Silencing BCL-2 promoted tumor cell apoptosis [17], and activating RIG-I stimulated the immune system to destroy the tumor through a mechanism that involved induction of type I IFN by RIG-I [18].These results suggest that single or combinatorial therapies that exploit the ability of RIG-I to stimulate beneficial immune responses may have potentially useful therapeutic applications for the treatment of melanoma and perhaps other types of cancer.
In order to reconcile this paradox, we propose that RIG-I might be a double-edged sword between inflammation and cancer by stimulating defensive immune responses and inducing infected cells into apoptosis (Figure 1). Indeed, recent data show that the RIG-I signaling pathway is involved in inflammasome-associated caspase-I activation and apoptosis of virus-infected cells [19-21]. RIG-I acts as a sensor for inflammasome activation, leading to Caspase-I activation and generation of mature IL-1β (IPS-1-independent RIG-I signal transduction) [16,22].
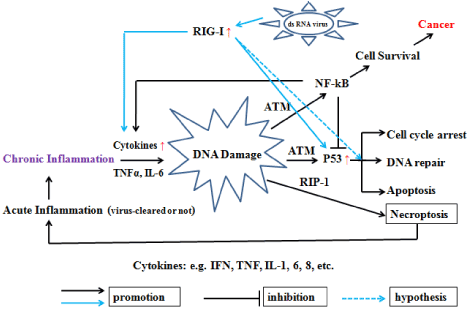
Figure 1. Simplified diagram showing RIG-I potential role between inflammation and cancer If not cleared timely by surrounding macrophages, virus-infected acute inflammatory cells will gradually become chronic inflammatory cells, and these cells produce TNFα, IL-6 and other pro-inflammatory factors, and thus, combined with RIP-I or ATM activation by DNA damage, induce infected cell into necroptosis or activate NF-kB and p53 signaling pathways to regulate cell survival or death, respectively. Programmed necroptosis usually results in acute inflammation to clear virus, while activation of NF-κB typically promotes cell survival by making cells resistant to apoptosis and stimulating cell growth. On the other hand, p53 activation is growth inhibitory and, depending on the cell type and the stress type, can result in temporary cell cycle arrest, irreversible arrest (senescence), or apoptosis. These cardinal differences in the outcomes of p53 and NF-κB activation are determined by the nature of the genes that they modulate as transcription factors. NF-κB activates transcription of positive growth regulators, various antiapoptotic factors, and secreted attractants of the immune response (cytokines and chemokines). p53, however, induces genes that encode cell cycle checkpoint regulators, pro-apoptotic factors, and secreted growth inhibitors. Consistent with these functional differences, p53 and NF-κB are deregulated in opposite directions in tumors. Although p53 is a tumor suppressor that is commonly inactivated, NF-κB behaves like an oncogene, showing constitutive activation in the majority of cancers. dsRNA Viruses can induce the expression of RIG-I, further stimulate the expression and secretion of IFNs, TNFα, IL-6 and other cytokines, in the meantime, up-regulate p53 expression. In the present hypothesis, RIG-I functions as an anti-oncogene to arrest cell cycle for DNA repair or apoptosis induction directly.
Overall, NF-κB activation and establishment of inflammation create a situation in which p53 can no longer effectively exert its function as an eradicator of transformation-prone cells. The severity of the consequences that follow depends dramatically on the duration of inflammation. For “typical” acute infections, strong NF-κB activity lasts from a few hours to a few weeks. No association has been found between occurrence of diseases associated with acute inflammation and cancer. Chronic inflammation, however, is a completely different story since the cells in inflamed tissues may exist in an NF-κB-activated/ p53-suppressed state for amounts of time sufficient for the acquisition of genetic and epigenetic alterations supporting advanced transformed phenotypes. This makes chronic inflammation a dangerous physiological condition that is functionally equivalent to a p53-suppressing oncogene. Activation of NF-κB functionally leads to “weakening” of p53 activities and phenotypically resembles partial deficiency of p53. Different aspects of p53 function are repressed by NF-κB to different degrees. RIG-I plays a two-edged sword role between inflammation and cancer by regulating nuclear transcriptional factors NF-κB and p53.
In short, RIG-I may possess tumor-suppressor properties through its ability to activate IFN responses, arrest cell cycle, delay cell growth and induce cell apoptosis.
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, et al. (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730-737. [Crossref]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, et al. (2006) 5'-Triphosphate RNA is the ligand
for RIG-I. Science 314: 994-997. [Crossref]
- Matsumiya T, Stafforini DM (2010) Function and regulation of retinoic acid-inducible gene-I. Crit Rev Immunol 30: 489-513. [Crossref]
- Cui Y (2008) Progress of The Research on Retinoic Acid Inducible Gene I. Prog Biochem Biophy 35: 380-386.
- Cho YS, Challa S, Moquin D, et al. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112-1123. [Crossref]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, et al. (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A 104: 582-587. [Crossref]
- Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7: 211-217. [Crossref]
- Kundu JK, Surh YJ (2008) Inflammation: gearing the journey to cancer. Mutat Res 659: 15-30. [Crossref]
- Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, et al. (2004) Gastric cancer originating from bone marrow-derived cells. Science 306: 1568-1571. [Crossref]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, et al. (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285-296. [Crossref]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, et al. (2004) NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431: 461-466. [Crossref]
- Arnott CH, Scott KA, Moore RJ, Hewer A, Phillips DH, et al. (2002) Tumour necrosis factor-alpha mediates tumour promotion via a PKC alpha- and AP-1-dependent pathway. Oncogene 21: 4728-4738. [Crossref]
- Kamp DW Shacter E, Weitzman SA (2011) Chronic inflammation and cancer: the role of the mitochondria. Oncology (Williston Park) 25: 400-410, 413. [Crossref]
- Pejovic T, Nezhat F (2011) Missing link: inflammation and ovarian cancer. Lancet Oncol 12: 833-834. [Crossref]
- Wallach D, Kovalenko A, Kang TB (2011) 'Necrosome'-induced inflammation: must cells die for it? Trends Immunol 32: 505-509. [Crossref]
- Kumagai Y, Akira S (2010) Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol 125: 985-992. [Crossref]
- Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, et al. (2008) 5'-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med 14: 1256-1263. [Crossref]
- Petrocca F, Lieberman J (2008) RIG-ing an antitumor response. Nat Med 14: 1152-1153. [Crossref]
- Li K, Chen Z, Kato N, Gale M Jr, Lemon SM (2005) Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J Biol Chem 280: 16739-16747. [Crossref]
- Foy E, Li K, Sumpter R Jr, Loo YM, Johnson CL, et al. (2005) Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A 102: 2986-2991. [Crossref]
- He L, Chen Y, Wu Y, Xu Y, Zhang Z, et al. (2017) Nucleic acid sensing pattern recognition receptors in the development of colorectal cancer and colitis. Cell Mol Life Sci. [doi: 10.1007/s00018-017-2477-1] [Crossref]
- Poeck H, Ruland J (2012) From virus to inflammation: mechanisms of RIG-I-induced IL-1β production. Eur J Cell Biol 91: 59-64. [Crossref]
2021 Copyright OAT. All rights reserv