Abstract
Within the last decades, scientists have gained detailed insight into mechanisms of synaptic transmission, hyperexcitability, excitotoxicity and neurodegeneration. These (patho) physiological processes are substantially regulated by voltage-gated Ca2+ channels (VGCCs), the glutamatergic system and trace metal ions such as Zn2+ and Cu2+. Whereas early studies in the field provided significant but only fragmentary insight, recent findings enable us to understand the complex crosstalk between individual voltage- and ligand-gated ion channels entities and divalent heavy metal ions in the brain. Dysbalance in Ca2+, Zn2+ and Cu2+ homeostasis and also the glutamate system may be linked to the pathogenesis of neurodegenerative disorders and hyperexcitability-related disease states, such as Alzheimer’s disease and epilepsy. Starting from hippocampal CA3 mossy fiber terminals, we create an integrative overview of the complex functional and structural interplay of Ca2+ channels, the glutamate system and trace metals.
Key words
copper, zinc, kainate, NMDA, R-type, T-type
Abbreviations
: [Ca2+]i: free cytosolic Ca2+ concentration; AD: Alzheimer’s disease; 4-AP: 4-aminopyridine; AMPA: a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CNS: central nervous system; DAG: diacylglycerole; DHP: dihydropyridine; EC: excitation-contraction; ER: endoplasmatic reticulum; ES: excitation-secretion; HVA: high-voltage activated; KA: kainic acid; LVA: low-voltage activated; LTP: long-term potentiation; mGluR: metabotropic glutamate receptor; NMDA: N-methyl-D-aspartate; PKC: protein kinase C; PLC: phospholipase C; RTN: reticular thalamic nucleus; SR: sarcoplasmatic reticulum; VGCC: voltage-gated Ca2+ channels
Introduction
Voltage-gated Ca2+ channels (VGCCs) are central players in mediating Ca2+ influx into living cells. Ten different Cav-a1 subunits of VGCCs complexes have been cloned so far, each exhibiting specific electrophysiological and pharmacological properties [1]. Within the central nervous system (CNS) there is a time- and regional specific distribution pattern of VGCCs that significantly contributes to the establishment of brain eurhythmia. Using electrophysiological, molecular and immunolocalisation techniques we now know that VGCCs are differentially distributed throughout the neurolemma, some of them specifically mediating synaptic transmission and long-term potentiation (LTP). Within the last decade it turned out that within the synaptic fusion machinery also divalent heavy metal, i.e. Zn2+ and Cu2+ ions play a major role and exert complex effects on both pre- and postsynaptically localized voltage- and ligand-gated ion channels [2]. Importantly, it turned out that glutamate receptors build up a functional triade with specific VGCC entities as well as Zn2+ and Cu2+ ions and that this functional interdependence is of major relevance for the initiation and perturbation of neural circuitry specific rhythmicity. In consequence, alteration in this sophisticated system is to result in central dysrhythmia leading to aberrant brain excitability or cognitive impairment as observed in dementia. Though we know that the functional interplay between the calcium system, the glutamate system and divalent heavy metal ions severely influences neural excitability it remains unclear how a dysbalance in these systems is to trigger hyperexcitability and ictogenesis.
In this review we provide detailed information on structure and function of VGCCs, their differential distribution within the CNS and their electrophysiological modification by divalent heavy metal ions. Then, we functionally integrate VGCCs and Zn2+/Cu2+ into the glutamate system to elaborate how these players can interfere and dysfunction contributing to epilepsy or dementia disorders phenotype.
Structural, functional and pharmacological characteristics of voltage-gated Ca2+ channels
Voltage-gated Ca2+ channels are of central relevance in mediating Ca2+ influx into living cells. They can trigger various physiological processes such as excitation-contraction (EC) coupling [3], excitation-secretion (ES) coupling [4], hormone and transmitter release [5,6] and regulation of gene expression [7,8]. Structurally, VGCC complexes are heteromultimeric assemblies composed of a central pore-forming, ion-conducting Cav-α1 subunit and various auxiliary subunits (α2-δ(1-4), b(1-4) and γ(1-8)) (Figure 1). Ten different Cav-α1 subunits have been cloned which can be differentiated according to their electrophysiological and pharmacological properties into high-voltage activated (HVA) and low-voltage activated (LVA) channels. High-voltage activated Ca2+ channels are further subdivided into dihydropyridine (DHP) -sensitive L (“long-lasting”)-type Cav1.1–1.4 and less DHP-sensitive non-L-type Cav2.1–2.3 channels. The LVA T-(“transient/tiny”) type comprises the Cav3.1-3.3 Ca2+ channels [1,9,10]. These channels are activated at rather negative membrane potentials (Va = (-44) – (-46) mV, ta = 1 - 7 ms at -10 mV), exhibit fast inactivation (Vh = (-72) – (-73) mV, th = 11 – 69 ms at -10 mV), and small single-channel conductance (7.5 - 11 pS) [1, 10-14]. By contrast, HVA L- and non-L-type channels are activated at much stronger depolarization (e.g. Cav1.1: Va = 8 – 14 mV, ta > 50 ms at +10 mV) [15], display higher single-channel conductances (L-type: 13 - 25 pS; Non-L-type: 9 – 20 pS), and prolonged-channel opening compared to T-type channels (Cav1.2: tslow = 1100 ms; Cav1.3: tslow = 1700 ms) [1, 16]. One must note, however, that Cav1.3 L-type Ca2+ channels were also shown to exhibit mid-voltage activating characteristics under special physiological and electrophysiological conditions (Va = (-15) – (-37) mV, ta <1 ms at +10 mV) [17-21].
Pharmacologically, L-type Ca2+-channels are characterized by high sensitivity towards DHPs, e.g. nifedipine (Cav1.1: IC50 = 0.15 nM at -65 mV, nitrendipine, nisoldipine, nicardipine); phenylalkylamines, e.g. verapamil, gallopamil, devapamil (Cav1.2: IC50 = 50 nM at -60 mV) and benzothiazepines, e.g. diltiazem (Cav1.2: IC50 = 33 mM at -60 mV) [18,22-25]. Whereas the first DHPs to be developed exerted both cardiac and vascular effects, next-generation DHPs predominately target vascular smooth muscles leading to relaxation and thus antihypertensive action [26]. An experimental activator of L-type channels is BayK8644, which is not used in clinical applications.
High-voltage activated non-L-type channels which are predominately engaged in synaptic transmission in the brain are effectively blocked by various snail and spider toxins: Ω-agatoxin IVA, derived from the funnel web spider Agelenopsis aperta targets Cav2.1 (IC50 = 1 - 3 nM), ω-conotoxin GVIA derived from Conus geographus blocks Cav2.2 (IC50 < 30 nM) and ω-conotoxin MVIIC 2, a toxin from the venom gland of marine snail Conus magnus, targets both Cav2.1 (IC50 > 7 nM ) and Cav2.2 Ca2+ channels (IC50 > 100 nM) [1,27-30]. For a long time, Cav2.3 R-type channels were considered to be unique as they turned out to be resistant to most Ca2+-channel blockers outlined above. However, in 1998, the spider peptide toxin SNX-482, derived from the venom of the tarantula Hysterocrates gigas, was shown to be a selective Cav2.3 channel inhibitor at low nanomolar concentrations (IC50 = 15-30 nM) [31]. In addition, Cav2.3 turned out to be highly sensitive to Ni2+ (IC50 = 27 mM), a property they share with Cav3.2 Ca2+ T-type channels (IC50 = 5-10 μm; [32]. Although these naturally derived toxins are predominantly of experimental interest since they are not applicable in humans, Cav2.1-2.3 VGCCs turned out to serve more and more as potential target in epilepsy and pain treatment. Gabapentin, for example, inhibits Cav2.1 channels via interaction with the a2-d auxiliary subunits (albeit on-selectively), and it can influence epilepsy and pain in humans [33]. Ziconotide (ω-conotoxin MVIIA, i.e. SNX-111), a toxin derived from the marine piscivorous snail Conus geographus, is likely to inhibit Cav2.2 Ca2+ channels and is a potent drug in humans who turned out to be refractory or non-tolerant to opioids (IC50 = 55 pM) [29]. For the LVA Ca2+ channels, various blockers have been proposed, including the tetraline derivative mibefradil (Cav3.1: IC50 = 270 nM; Cav3.2: IC50 = 140 nM), the scorpion toxin kurtoxin (IC50 = 15 nM) [34], as well as Ni2+-ions (Cav3.1: IC50 = 250 aM; Cav3.2: IC50 = 12 aM: Cav3.3: IC50 = 216 aM) [1,10,34,35]. Recently, azetidinones and spiro-azetidines have been described as novel potential blockers of the T-type Ca2+ channel Cav3.2 for the treatment of neuropathic and inflammatory pain [36]. However, none of these potential T-type blockers has reached clinical application so far.
Diphenylalkylamine derivatives such as flunarizin or cinnarizin exhibit a non-specific blockade on VGCCs. Clinically, these drugs are used for dilatation of cerebral arteries to enhance cerebral blood flow.
Given the important functional implications of VGCCs, it is not surprising that a number of voltage-gated Ca2+-channelopathies have been identified so far. An overview of the tissue distribution, pharmacological and functional aberrations of the individual Cav-α1 subunits is given in Table 1.
Cav-α1 |
Pharmacology |
Tissue affected |
Syndromes associated |
Cav1.1 |
Dihydropyridne,
Benzothiazepine,
Phenylalkylamine,
TaiCatoxin,
Calciseptine
Calcicludine, FS-2 |
skeletal muscles |
Hypokalemic periodic paralysis type 1 (HypoPP1), malignant hyperthermia type 5 (MHS5) |
Cav1.2 |
ubiquitary |
Timothy syndrome (LQT8, epilepsy) |
Cav1.3 |
ubiquitary |
not known |
Cav1.4 |
retina |
x-linked conginital stationary night blindness 2 (xCSNB2), X-linked cone-rod dystrophy type 3 (CORDX3) |
Cav2.1 |
ω-Agatoxin IVA |
CNS |
Absence-epilepsy, episodic ataxia type 2, spinocerebellar ataxia type 6, familial hemiplegic migraine, Lambert-Eaton myastenie-Syndrome |
Cav2.2 |
ω-Conotoxin GVIA |
CNS/PNS |
Lambert-Eaton myastenie-syndrome |
Cav2.3 |
SNX-482, Ni2+ |
CNS/PNS |
not known |
Cav3.1 |
Mibefradil,
Kurtoxin,
Ni2+ |
CNS/PNS |
not known |
Cav3.2 |
CNS/heart |
Absence-epilepsy (CAE), Autism spectrum disorders (ASD) |
Cav3.3 |
CNS |
not known |
Table 1. Pathophysiological implications of voltage-gated Ca2+ channels in channelopathies. VGCCs are subdivided into HVA L- and Non-L-type and LVA Ca2+ channels based on activation characteristics and the pharmacological inhibition profile (modified from [145].
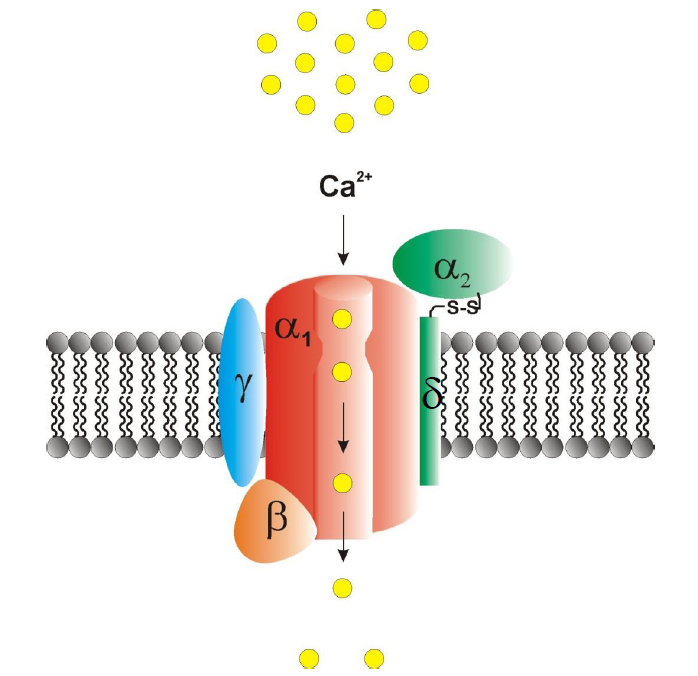
Figure 1. Structure of heteromultimeric voltage-gated Ca2+ channels complexes. The pore-forming Cav-α1 subunit is a four domain macromolecular protein that can harbour extracellularly located trace metal binding sites. Various auxiliary subunits, i.e. the α2δ, β and γ subunit, are capable of modulating basic electrophysiological and pharmacological properties of the Cav-α1 subunit. Mutations in both pore-forming and auxiliary subunits are related to voltage-gated Ca2+ channelopathies (Table 1) (adapted from [145]).
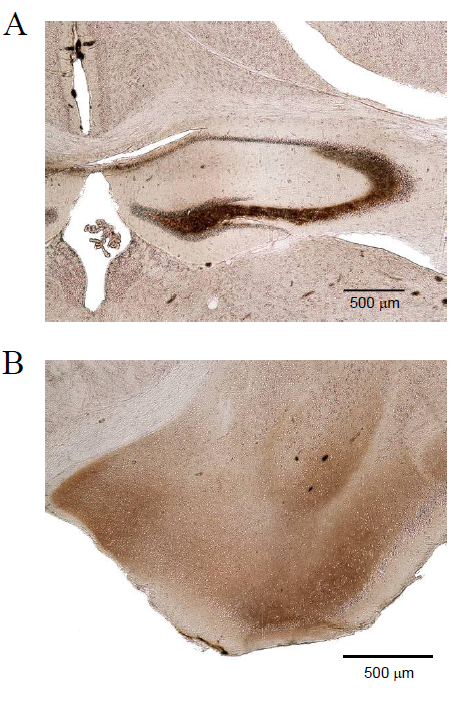
Figure 2. Distribution of Zn2+ in the normal mouse brain. TIMM staining of a coronal cryosection of a mouse brain (C57BL/6). Note that Zn2+ accumulations are predominantly localized in the hippocampus (A), amygdala (B), striatum and cortex. Hp, hippocampus, CA, cornu ammonis, DG, dentate gyrus, bar: 500 μM.
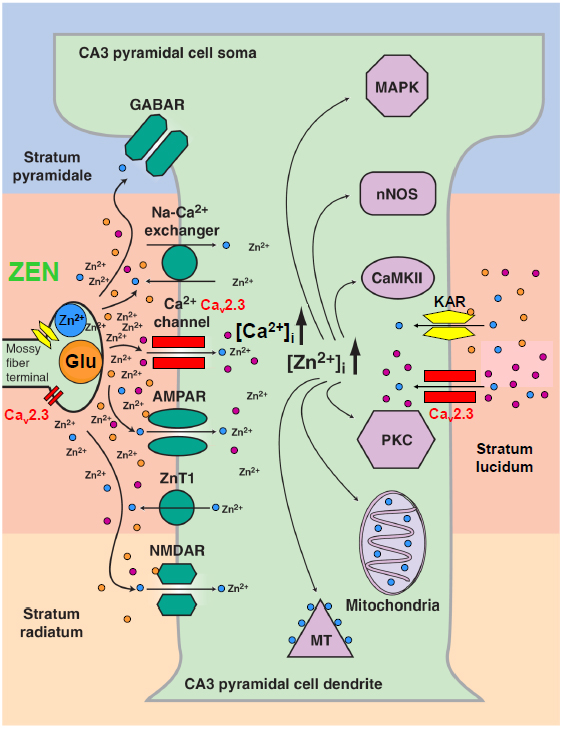
Figure 3. Physiological interdependence of VGCCs (Cav2.3), the glutamate system and divalent heavy metal ions (Zn2+). The hippocampal mossy fiber terminal is an example of the complex interaction of VGCCs with glutamate and Zn2+. Presynaptically, various VGCCs such as Cav2.1, Cav2.2 and Cav2.3 are expressed. Cav2.3 channels were shown to be involved in presynaptic long-term potentiation. The mossy fibre terminal represents a gluzincerigc synapse in which glutamate and Zn2+ can be released into the synaptic cleft with high local concentrations. Sub- and postsynaptically, glutamate can exert its action directly on ionotropic (NMDA) glutamate receptors (iGluR) and metabotropic glutamate receptors (mGluR). Importantly, glutamate can serve as a chelator that reduces the activity of free Zn2+ and results in complex modulation of both pre- and postsynaptically localized Cav2.3 Ca2+ channels (modified from [127]).
It is noteworthy that auxiliary subunits (α2δ1-4, β1-4 and γ1-8) are capable of substantially modulating basic electrophysiological and pharmacological properties as well as plasma level expression of the Cav-α1 subunits [5,37].
Under physiological conditions, Ca2+ influx into intact neurons is highly organized in space, frequency and amplitude as the spatiotemporally integrated free cytosolic Ca2+ concentration [Ca2+]i contains specific information [38]. Principally, Ca2+ can enter the cytosol via the Na+/Ca2+ exchanger, through release from intracellular stores, e.g. ER and SR, via VGCCs and an armamentarium of other potentially less-specific voltage- and ligand gated cation channels. VGCCs however provide a powerful mechanism to directly link neuronal activity to Ca2+ influx. Until buffering mechanisms restore the resting Ca2+ levels [39,40], [Ca2+]i regulates critical cellular functions, including channel modulation, neurotransmitter release and gene transcription. This Ca2+ influx is also supposed to be of central relevance in hyperexcitability and excitotoxicity mediated neurodegeneration. For example, the Ca2+ hypothesis of epileptogenesis claims that alterations in [Ca2+]i may play a crucial role in the development of epilepsy [41-43]. High- and low-voltage activated Ca2+ channels are likely to be predominant mediators of [Ca2+]i elevation during most epileptiform activity [41,44]. In hippocampal neurons, Ca2+ current density was up-regulated during epileptogenesis [45] and inhibition of VGCCs effectively depressed epileptiform activity [46-48]. Ca2+ channels were further shown to be of central relevance in mediating potential epileptiform activity on the cellular electrophysiological level, including phenomena such as afterdepolarization, plateau potentials or exacerbation of low-threshold Ca2+ spikes/rebound burst firing thus mediating seizure initiation, propagation and kindling [34, 49-53]. Concomitant, VGCCs exert major effects in excitotoxicity and neurodegeneration by contributing to the devastating pathophysiology of human neuronal diseases associated with neurodegeneration [54, 55] (Table 1). Thus, blockade of VGCCs turns out to be a reasonable approach to pharmacologically interfere with seizure activity, excitotoxicity and neurodegeneration [56-61].
In this context, interaction partners of VGCCs turned out to be most relevant in drug discovery and development. Recently, an exceptional study on quantitative proteomics of Cav2 channel nano-environments, using knockout-controlled multiepitope affinity purifications together with high-resolution quantitative mass spectroscopy was carried out to unravel the molecular players in local subcellular signalling [62]. About 200 proteins have been identified that clearly differ in abundance, stability of assembly and preference for the individual Cav2 subunits. These potential interaction partners included kinases and phosphatases, cytoskeleton proteins, enzymes, SNAREs, modulators and small GTPases, various G-protein coupled receptors, ion channels and transporters, adaptors, extracellular matrix proteins, cytomatrix components, protein trafficking components and additional proteins of yet unknown function.
The glutamate system and its interaction with VGCCs
One of the most intensively studied CNS structures in terms of neuronal hyperexcitability and excitotoxicity mediated neurodegeneration is the hippocampus, where along with other ion channel entities, VGCCs are highly expressed. Physiological neuronal computation in the brain requires a well-tuned balance between ongoing excitatory transmission and controlled inhibition. Excessive excitatory activity alone results in neuronal damage and cell death through mechanisms known as excitotoxicity [54,55]. The glutamate system and VGCCs are key players in excitotoxicity and hyperexcitability [63]. L-glutamate acts on both metabotropic glutamate receptors (mGluR) and ionotropic glutamate receptors, namely N-methyl-D-aspartate (NMDA)-, a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)- and Kainic acid (KA)-receptors. Kainic acid is a well-characterized excitotoxin derived from Digenea simplex. In the hippocampus, KA induces degeneration predominantly of CA3 pyramidal neurons and hyperexcitability in surviving pyramidal cells thus triggering complex partial seizure activity [64]. Alternatively spliced KA-receptors, e.g. GluR5-7 and KA1-2, are widely distributed in the hippocampal formation on somata, dendrites, neurites and synapses. They exert various effects including regulation of glutamate release [65], modulation of postsynaptic currents [66], and regulation of GABAergic (gabazincergic) synaptic transmission in the hippocampus [67]. Both NMDA- and KA-receptor activation is further associated with activation of VGCCs due to prolonged depolarisation and Ca2+-mediated excitotoxicity which might in part be responsible for neuronal cell death [68]. However, the exact mechanisms yet have to be explored. Interestingly, KA-Rs are not only expressed postsynaptically at hippocampal CA3 dendritic spines but also presynaptically at mossy fibre terminals within the stratum lucidum (Figure 2). Whereas single fibre activation causes negligible KA-Rs responses postsynaptically, repetitive activation of mossy fibres results in greatly enhanced KA-R activation. Due to the differences in AMPA- and KA-receptor current kinetics, the resulting excitatory postsynaptic potentials exhibit two components. Interestingly, KA-receptors are activated already by quantal glutamate release into the peripheral cleft. They can induce tonic depolarisation, thereby bringing cells closer to the activation threshold and enhancing the relative importance of single inputs [68]. Presynaptically, KA-receptors act as auto-receptors that sense the glutamate released. These KA-Rs mediated responses exhibit an intriguing multidimensional regulation pattern: 1. dose-dependent effects (Schmitz et al., 2001a; Schmitz et al., 2001b) with a biphasic, bidirectional modulation of glutamate release: low KA concentrations facilitate glutamate release and enhance synaptic currents [69] whereas high KA levels cause inhibition of glutamate release and reduction of synaptic current. 2. time-dependent effects of KA-receptors mediated action with early stimulatory effects on neurotransmission and late depressing effects. There is intense and on-going discussion on how KA-receptors mediate these multiple spatiotemporal presynaptic effects. Low concentrations might result in only small depolarisations, whereas higher concentrations cause presynaptic inhibition of neurotransmission due to ionotropic action of KA-receptors and strong depolarisations that inhibit Na+- and Ca2+-channels [69-71]. Recent studies clearly indicate that presynaptic KA-receptors substantially contribute to long-lasting afterdepolarisations. They can, therefore, modulate the amount of presynaptic Ca2+ contributing to frequency-dependent facilitation of glutamate release [69].
Both VGCCs and KA-receptors modulate neurotransmission and synaptic long-term plasticity in the hippocampus probably related to the profound excitotoxic effects of KA in this brain region as well [69,72,73]. Interestingly, gene profiling studies using microarray analysis revealed that various neuroprotectants and neurotransmitters are up-regulated in a rat KA seizure model [74], some of which clearly interact with or functionally modulate VGCCs, e.g. hsp70 [75] and neurokinin 1 (NK1) for Cav2.3 Ca2+ channels [76] or interfere with other VGCCs. Additionally, other factors such as metallothionein-1 and -2, which indirectly affect VGCCs, e.g. via interference with Zn2+-homeostasis [2] were also up-regulated in the KA seizure model [74]. Up to now little is known about the functional role of VGCCs in neurodegeneration, such as Alzheimer’s disease, although some studies point to a neuroprotective effect of Ca2+ channel blockers [34,50,57,77]. In hippocampal neurons, L-type VGCCs, in addition to NMDA-receptors, are known to play a role in excitotoxic processes [78]. Furthermore, mGluR regulation of VGCCs, e.g. N-type channels, was shown to limit NMDA mediated toxicity in the neostriatum [79]. Blocking L- and N-type VGCCs is effective in neuroprotection following traumatic cell injury as well [80].
Recently, Zaman et al. [51] demonstrated that Cav2.3 Ca2+ channels and a Ca2+-activated K+-channel, i.e. SK2, can form a functional microdomain that mediates slow afterhyperpolarisations (sAHPs) in reticular thalamic nucleus (RTN) neurons following low-threshold Ca2+-spikes. This mechanism is capable of sustaining oscillatory burst activity in RTN neurons and consequently, Cav2.3-/- mice exhibit reduced burst charges and suppressed sAHP. T-type Ca2+ channels per se did not seem to be sufficient to maintain Ca2+ levels that can trigger sAHP, the latter however is a prerequisite for repriming T-type Ca2+ channels and sustained rebound bursting [51]. Interestingly, this functional scenario of Cav2.3 mediated SK2 activation has previously been described in the CA1 region as part of a functional triad including NMDA-receptors, Cav2.3 and SK2. Similar to RTN neurons, Cav2.3 and SK2 channels induce AHPs that are likely to serve as a negative feedback in regulating synaptic activity and plasticity in dendritic spines [81-83], as well as epileptogenicity [84]. However, the spatio-temporal organisation of Ca2+-channel and the mGluR interaction is rather complex. Studies by Metz et al. [85] suggested that AHPs that typically follow action potentials in CA1 pyramidal neurons are mediated by a Ni2+-sensitive current, the pharmacological and biophysical profile of which clearly resembles Cav2.3 Ca2+currents. As AHPs are directly related to burst firing in CA1 neurons, Cav2.3 R-type currents are relevant for encoding hippocampal place fields and enhancement of synaptic plasticity. Further studies revealed that group I mGluR1 and group V mGluR5 can dramatically alter firing patterns of CA1 pyramidal neurons via a complex, activity-dependent modulation of Cav2.3 R-type Ca2+ channels (Park et al., 2010).
Electrophysiological studies in rat hippocampal CA1 neurons elucidated that VGCCs such as Cav2.3 R-type Ca2+ channels can also trigger epileptiform activity by contributing to plateau potential generation following muscarinic M1/M3-R stimulation via a Gq/11, phospholipase C (PLC), diacylglycerole (DAG) and protein kinase C (PKC) mediated signalling pathway [52,53]. Consistently, seizure susceptibility studies in Cav2.3-/- mice exhibited a dramatic resistance to both generalized tonic-clonic and KA-induced hippocampal seizures and increased resistance to KA-induced hippocampal cell loss [86-88]. However, up to now we still lackdetailed information on the functional implications of VGCCs in neurodegeneration itself. The molecular chaperone hsp70 functionally interacts with the II-III loop of the Cav2.3 R-type VGCC and thus might also regulate PKC effects on this channel [75]. Besides, KA-receptors also exert metabotropic effects, e.g. via a pertussis-toxin sensitive G-protein coupled signaling pathway including PLC, DAG and PKC thus providing a possible “crosstalk” with pre- and postsynaptically localized VGCCs which are strongly Ca2+- and PKC-regulated [67,89]. It has further been elucidated that mutations in EFHC1, a novel C-terminal interaction partner of the Cav2.3 VGCC, cause juvenile myoclonic epilepsy in humans, indicating that Cav2.3 is likely to be involved in neuronal apoptotic processes related to excitotoxicity [90]. A critical role of Cav2.3 in neuronal degeneration is supported by the observation that antiepileptic drugs known to block Cav2.3, e.g. topiramate and lamotrigine, also exert strong neuroprotective effects [87]. Other VGCCs have been implicated in the pathogenesis of epilepsies and neurodegeneration as well. Cav3.2 T-type Ca2+ channels have been related to hippocampal hyperexcitability and neuronal cell death in the rat pilocarpine model of limbic seizures [49,91]. The individual contribution of Cav2.3 R-type and Cav3.2 T-type Ca2+-channels to hippocampal hyperexcitability and neurodegeneration is likely to be dependent on the different experimental paradigms that have been used, e.g. acute, pharmacologically induced generalized tonic-clonic seizure models (4-AP) or hippocampal seizure models (KA, NMDA) versus pharmacologically induced status-like hippocampal seizure models following pilocarpine administration. Calcium influx via Cav1.2 L-type and Cav2.1 has also been related to neurodegenerative processes. Missense gain-of-function mutations within the Cav1.2 a1-subunit result in the Timothy-syndrome, a multisystem disorder characterized by a plethora of organ dysfunctions including lethal arrhythmias, congenital heart disease, syndactyly, immune deficiency and intermittent hypoglycaemia [92]. Interestingly, the Cav1.2 gain-of-function mutations associated with impaired channel inactivation are associated with various neuropsychiatric syndromes, such as autism spectrum disorders, intellectual disability and epilepsy [92,93].
Gain-of function mutations of neuronal Cav2.1 Ca2+ channels are known to be involved in the etiopathogensis of familial hemiplegic migraine type 1(FHM1). The Cav2.1 channel is expressed in various brain stem nuclei, the cerebral cortex and terminal ganglia and closely related to the control of nociception. Various missense mutations have been reported for the gene encoding for the pore-forming subunits of Cav2.1. Gain-of-function mutations of Cav2.1 channels were studied in detail using FHM knock-in mouse models displaying increased neurotransmitter release from cortical neurons [94,95]. Glutamate release on pyramidal neurons is hypothesized to facilitate and propagate experimentally induced cortical spreading depression (CSD) that underlies the phenomenon of migraine aura [96].
Zinc and copper – divalent heavy metal ions in the central nervous system
In pharmacology, heavy metal ions are widely used to characterize and distinguish individual components and subtypes of GluRs and VGCCs. Many of these heavy metal ion entities turned out to be of merely theoretical, i.e. pharmacological interest, with two major exceptions: Zn2+ and Cu2+ ions. Within the last decade, Zn2+ emerged to be one of the most important heavy metal ions within the CNS, to the extent that is was sometimes referred to as “the calcium of the twenty-first century” [2]. Some forebrain neurons, i.e. zinc-enriched neurons (ZEN) exhibit extremely high Zn2+-concentrations, e.g. glutamatergic (gluzinergic) neurons of the amygdala, the cerebral cortex and particularly the hippocampus. Zinc-storing vesicles, so called zincosomes exist in both GABAergic (gabazinergic) neurons, e.g. in the cerebellum and glutamatergic (gluzinergic) neurons, e.g. in granule cell mossy fibres boutons [2] with Zn2+-transporters such as ZnT3 and ZnT4 being responsible for the strong vesicular Zn2+-enrichment of up to 350 mM [2, 97]. Plasma Zn2+-concentrations are about 15 mM, with free Zn2+ in the nanomolar range [97,98]. In the brain, about 90 % of Zn2+ is bound to metalloproteins and 10% in vesicles [97]. Like Zn2+, Cu2+ also accumulates in the CNS, predominantly in adrenergic and glutamatergic (glucupergic) neurons in the hippocampus, the olfactory bulb and the locus coeruleus [99-101]. Normal extracellular Cu2+-concentrations are in the range of 0.2 – 1.7 mM [98] and are frequently increased in neurodegenerative processes such as Alzheimer’s disease, then reaching levels of up to 200 – 400 mM. Both divalent trace metals, Zn2+ and Cu2+, are implicated in a range of neurological disease states in humans that are characterized by alterations in neuronal excitability and/or neurodegeneration. For example, there is strong evidence of a pathogenic Zn2+- and Cu2+-interaction with amyloid-b in Alzheimer’s disease. As an established pathogenetic process, pathological accumulation of Cu2+ can lead to Wilson’s disease, i.e. hepatolenticular degeneration. In addition, alterations of Zn2+-metalloproteins, such as superoxide dismutase or metallothionein, can cause amyotrophic lateral sclerosis (ALS, Lou Gehrig’s disease) [2].
Furthermore, Zn2+ is known to exert effects on epileptic activity and excitotoxicity [102]. The role of Zn2+ and Cu2+ in epilepsy and excitotoxicity is complex, and partially ambivalent. Whereas a number of studies illustrate that Zn2+ is a potential ionic mediator of selective neuronal injury [103-106], others provide strong evidence that Zn2+ is a powerful neuroprotector [2,107-110]. Similarly, Zn2+ was reported to serve as both a proconvulsant [111] and anticonvulsant [112,113] in humans and various animal models. These findings further support the apparent „Janus”-like behaviour of Zn2+ ions in modulating neurodegeneration and seizure susceptibility.
However, most of these prima facie contradictory observations described in the literature are based on differences in voltage- and ligand-gated ion channel expression within various neuronal cell types investigated, e.g. hippocampal interneurons versus pyramidal cells. Following KA-induced limbic seizures, hippocampal interneurons exhibit a dramatic increase in cytosolic Zn2+-concentration and cell death which is supposed to be due to mitochondrial dysfunction [105] and activation of specific Zn2+-signalling pathways [114]. Hippocampal interneurons were further reported to express Ca2+-permeable AMPA-receptors [115], and to release Zn2+ from mitochondria and other intracellular stores or metallothioneins [116]. Zn2+-levels turned out to be higher in interneurons compared to hippocampal pyramidal cells [117] due to differences in Ca2+-AMPA-receptor expression, Ca2+-buffering systems and differences in mitochondrial metabolism [118]. Compared to interneurons, CA3 pyramidal cells display only a moderate increase in internal Ca2+-levels after KA treatment [117]. Findings of Zn2+-release, intracellular Zn2+-accumulation and its effects on KA-seizure susceptibility and excitotoxicity are rather divergent as well. Whereas extracellular chelation of Zn2+ in one study neither affected hippocampal excitability nor seizure-induced cell death [119], studies by Takeda et al. illustrated that Zn2+ can clearly attenuate KA-induced limbic seizure activity and concomitant neurodegeneration in the CA3 region, or induce inverse effects, when being chelated extracellularly [107-110,120]. Thus, by complex modulation of the inhibition - excitation balance, Zn2+-homeostasis is crucial for both the induction of and the prevention of hyperexcitability-related seizure development and neurodegeneration.
The molecular targets of Zn2+ and Cu2+ have been elaborated in detail in the past. Zinc, in particular, was shown to exert important functions in synaptic transmission, e.g. via inhibition of NMDA- and GABA(A)-receptors, modulation of AMPA-receptors, inhibition of the GABA transporter 4 (GAT4), enhancement of Glycine-receptors response and, most importantly, blockade of VGCCs. Using a heterologous expression system with Cav1.2, Cav2.3 and Cav3.2, VGCCs were originally reported to be the most sensitive Ca2+ channels with IC50 values of 10.9 ± 3.4 mM, IC50 = 31.8 ±12.3 mM and 24.1 ± 1.9 mM, respectively [121]. Inhibition of low-voltage activated Ca2+-current by micromolar Zn2+ has further been reported in the rat aorta smooth muscle cells, but also in CA1 pyramidal neurons [122]. Importantly, Zn2+ can exert distinct and partially opposite effects on Cav3.1-3.3 T-type Ca2+ channels [123]. Whereas Cav3.2 Ca2+ channels were blocked by submicromolar Zn2+ concentrations (IC50 = 0.78 ± 0.07 mM), Cav3.1 and Cav3.3 Ca2+ channels turned out to be less sensitive to Zn2+ (IC50 = 81.7 ± 9.1 mM and IC50 = 158.6 ± 13.2 mM, respectively). Hence, Zn2+ can be used for the pharmacological distinction of different T-type Ca2+ channels.
On the electrophysiological level, different Zn2+ effects can be explained by subtype-specific modulation of Zn2+ acting on multiple binding sites of Ca2+ channels and altering gating mechanisms. As a possible allosteric modulator of Ca2+ channels, Zn2+ is responsible for a shift to more negative potentials of the steady-state inactivation curves of Cav3.1-3.3 T-type Ca2+ channels and the steady-state activation curve for Cav3.1 and Cav3.3 [123]. Furthermore, inhibitory effects of Zn2+ are use-dependent and strongly suggest preferential Zn2+ binding to the resting state of T-type Ca2+ channels. Inactivation kinetics for Cav3.1 and Cav3.3 were significantly slowed, but not for Cav3.2 VGCCs. Deactivation kinetics of Cav3.3 were also significantly slowed upon Zn2+ exposure whereas Cav3.1 and Cav3.2 tail currents remained affected. Increased Cav3.3 mediated Ca2+ current was observed after Zn2+ application resulting in increased duration of Cav3.3 mediated action potentials. Consequently, Zn2+ can apparently serve as an opener of Cav3.3 Ca2+ channels [123].
Most importantly, Zn2+ ions can exhibit not only different modulatory effects on various voltage- and ligand-gated ion channels, but also enter cells via different channels such as VGCCs, AMPA-, NMDA- and KA-receptors, particularly when neurons exhibit repetitive activation/hyperexcitability [2,106,124]. Thus, both Ca2+ and Zn2+ can serve as synaptic or transsynaptic second messengers with extracellular diffusion i.e. spill over effects at mossy fibre terminals for example, enabling complex heterosynaptic modulation. In line with this, synaptically released Zn2+ can effectively inhibit LTP presynaptically at the mossy fiber synapse [125]. These findings directly corroborate the critical role of Cav2.3 R-type Ca2+-channels, serving as a Zn2+ target in presynaptic LTP [73,121,126].
Cav2.3 channel activity is not only affected by Zn2+. The channel itself, in turn, is likely to regulate cellular Zn2+-influx in an excitability dependent fashion thereby controlling intracellular Zn2+-signalling. Zn2+-ions significantly influence signal transduction factors, such as PKC, CaMKII, mitogen-activated protein kinase (MAPK) and probably PKA [127]. PKC activity, for example, is modulated by both Ca2+ and Zn2+ ions in a dose dependent and interactive fashion. At low Ca2+-levels (5 mM), Zn2+ enhances PKC activity, whereas high Ca2+ (> 50 mM) results in a Zn2+-mediated inhibition of PKC. As PKC functionally interacts with VGCCs such as Cav2.3, these findings suggest a further regulatory mechanism via [Ca2+]i, PKC and finally Zn2+ [128]. At low cytosolic concentrations, neuroprotective Zn2+-effects have been related to Akt-phosphorylation and hsp70 upregulation [75,117,129] whereas the proapoptotic effect of Zn2+ is associated with caspase activation and release of proapoptotic proteins, such as cytochrome c or apoptosis-inducing factor (AIF) [2,130]. Interestingly, some of these factors are either Cav2.3 interaction partners, modulated by Cav2.3 or Zn2+-related proteins, or upregulated in the KA seizure model, e.g. MAPK, hsp70, neurokinin1 and metallothioneins [74]. Summarising the above findings, they suggest a complex crosstalk among VGCCs, Ca2+-, Zn2+- and Cu2+-ions, the exact nature of which, remains to be further understood. Given these findings, functional interactions among Zn2+, Cu2+ and Cav2.3 R-type channels became the focus of recent research efforts. T-type Ca2+ channels are exceptionally sensitive to low concentrations of Ni2+, Zn2+ and Cu2+ [123,131,132]. Nickel, a divalent cation has already been characterized as a selective blocker of Cav2.3 R-type, but also of Cav3.2 T-type Ca2+-channels. Recently, a key structural motif for Ni2+ and Zn2+ binding to Cav3.2 has been attributed to His191 located at the extracellular IS3-IS4 loop [32,132]. Sequence comparison revealed that two His-residues (H179 and H183) are present in the IS3-IS4 region of Cav2.3 Ca2+ channels as well. Following site-directed mutagenesis, electrophysiological studies revealed that both histidines are structural determinants of Ni2+ inhibition of the Cav2.3 R-type Ca2+ channel [133].
Recently, Sheglovitov et al. [134] carried out a series of exceptional experiments which may well revolutionize our knowledge on Zn2+ and Cu2+ effects on Cav2.3 Ca2+ channels. In vitro dose-concentration studies using HEK 293 heterologous expression systems and calibrated heavy metal ion concentrations revealed that Cav2.3 is a most sensitive target of Zn2+ and Cu2+ ions (IC50 = 1.3 ± 0.2 μM and IC50 = 18.2 ± 3.7 nM, respectively using voltage steps to -20 mV representative for effects on activation gating and IC50 = 8.1 ± 1.4 μM and IC50 = 269 ± 101 nM representative for action on conductance with voltage steps to +20 mV), in contrast to other channels and receptors, e.g. NMDAR (IC50 = 270 nM) and Cav3.2 (IC50 = 900 nM) [131,135].
Abolishing the effects on potential binding sites of divalent heavy metal ions by chelation or by substitution of key amino acid residues in the IS1–IS2 (H111) and IS3–IS4 (H179 and H183) loops substantially enhanced Cav2.3 mediated Ca2+ influx by shifting the voltage-dependence of activation toward more negative membrane potentials [136]. The authors further demonstrate that Cu2+ regulates the voltage dependence of Cav2.3 by affecting gating charge movements. The presence of Cu2+ resulted in slowing of gating charges transition into the “ON” position, delaying activation and reducing the voltage sensitivity of the channel. It was further shown that neurotransmitters, such as glutamate and glycine can serve as trace metal chelators by themselves and thus profoundly modulate activity of Cav2.3 Ca2+ channels by influencing their voltage-dependent gating.
Glutamate is released from presynaptic terminals and interferes with receptors on the pre- and postsynaptic membranes, conveying information between interconnected neurons. The spatiotemporal profile of glutamate action on direct and indirect targets is of high importance for proper signal transduction in the brain. Interestingly, glutamate substantially potentiated the activity of Cav2.3 channels at hyperpolarized potentials by shifting their voltage-dependent activation curve toward more negative voltages. Most importantly, the glutamate effect on Cav2.3 Ca2+ channels was clearly based on the chelating effect and mechanistically distinct from the activation of intracellular signal transduction cascades [137,138]. Glutamate exerts its action on Cav2.3 VGCCs from the extracellular side and although the trace metal binding character has been documented before [139,140] it was not considered to be physiologically relevant until now.
Considering the fact that the local concentration of glutamate in the synaptic cleft can transiently reach the millimolar range [141], all glutamate-sensitive postsynaptic channels and receptors should be considered as potential targets. The sensitivity of Cav2.3 Ca2+ channels to glutamate is based on the presence of trace metals in the extracellular milieu. Notably, the concentration of free or loosely bound Zn2+ and Cu2+ is elevated in the brain [142,143], and the release of these metals from synaptic vesicles in a voltage- and Ca2+-dependent fashion has been detected with various techniques [2].
It has never been reported before that the trace amounts of divalent heavy metals that often contaminate extracellular solutions [133,144] can exert tonic inhibitory effects on Cav2.3 voltage-dependent gating. Based on the observed shifts in IV-curves and changes in current kinetics in the presence of various Zn2+ and Cu2+ concentrations, Cav2.3 VGCC turned out to be mid-voltage activated in a Zn2+ and Cu2+ low/free environment.
It has been estimated that an average HEPES–TEA solution contains 50 nM Cu2+, which could be responsible for a 17 mV negative shift in the Cav2.3 activation curve. In addition, trace metal chelation also enhanced Cav2.3 current inactivation kinetics (Shcheglovitov et al., 2012). These findings are likely to have an impact on our view on Cav2.3 VGCCs, require a thorough re-assessment of previously reported electrophysiological studies on Cav2.3 channels and suggest that a plethora of new (patho)physiological function may exist for this channel entity.
Funding information
This work was funded by the Federal Institute for Drugs and Medical Devices, Bonn (Germany).
Competing interests
The authors declare no competing interests.
References
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J (2005) International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57: 411-425. [Crossref]
- Frederickson CJ, Koh JY, Bush AI (2005) The neurobiology of zinc in health and disease. Nat Rev Neurosci 6: 449-462. [Crossref]
- Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415: 198-205. [Crossref]
- Berggren PO, Yang SN, Murakami M, Efanov AM, Uhles S, et al. (2004) Removal of Ca2+ channel beta3 subunit enhances Ca2+ oscillation frequency and insulin exocytosis. Cell 119: 273-284. [Crossref]
- Catterall WA (2000) From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 26: 13-25. [Crossref]
- Catterall WA (1999) Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann N Y Acad Sci 868: 144-159. [Crossref]
- Bito H, Deisseroth K, Tsien RW (1997) Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol 7: 419-429. [Crossref]
- Hofmann F, Lacinová L, Klugba2021 Copyright OAT. All rights reservm channels: from structure to function. Rev Physiol Biochem Pharmacol 139: 33-87. [Crossref]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, et al. (2000) Nomenclature of voltage-gated calcium channels. Neuron 25: 533-535. [Crossref]
- Perez-Reyes E (2003) Molecular Physiology of Low-Voltage-Activated T-type Calcium Channels. Physiol Rev. 83: 117-161. [Crossref]
- Klöckner U, Lee JH, Cribbs LL, Daud A, Hescheler J, et al. (1999) Comparison of the Ca2 + currents induced by expression of three cloned alpha1 subunits, alpha1G, alpha1H and alpha1I, of low-voltage-activated T-type Ca2 + channels. Eur J Neurosci 11: 4171-4178. [Crossref]
- Klöckner, U., et al., (1999) Electrophysiological properties of structurally related T-type Calcium channel subunits expressed in stably transfected cell lines. Pflügers Arch-Eur J Physiol.
- Monteil A, Chemin J, Bourinet E, Mennessier G, Lory P, et al. (2000) Molecular and functional properties of the human alpha(1G) subunit that forms T-type calcium channels. J Biol Chem 275: 6090-6100. [Crossref]
- Monteil A, Chemin J, Leuranguer V, Altier C, Mennessier G, et al. (2000) Specific properties of T-type calcium channels generated by the human alpha 1I subunit. J Biol Chem 275: 16530-16535. [Crossref]
- Dirksen RT, Beam KG (1995) Single calcium channel behavior in native skeletal muscle. J Gen Physiol 105: 227-247. [Crossref]
- Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, et al. (2003) Cav1.4alpha1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J Neurosci 23: 6041-6049. [Crossref]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, et al. (2001) alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem 276: 22100-22106. [Crossref]
- Michna M, Knirsch M, Hoda JC, Muenkner S, Langer P, et al. (2003) Cav1.3 (alpha1D) Ca2+ currents in neonatal outer hair cells of mice. J Physiol 553: 747-758. [Crossref]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, et al. (2000) Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102: 89-97. [Crossref]
- Platzer J, et al (2000) Histological and physiological anomaliles in class D-L-type Ca2+ channel knockout mice. Naunyn-Schmiedeb. Arch Pharmacol 361: R70.
- Xu, W. and D. Lipscombe (2001) Neuronal Cav1.3alpha1 L-Type Channels Activate at Relatively Hyperpolarized Membrane Potentials and Are Incompletely Inhibited by Dihydropyridines. J Neurosci 21: 5944-5951.
- Hockerman GH, Johnson BD, Abbott MR, Scheuer T, Catterall WA (1997) Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels in transmembrane segment IIIS6 and the pore region of the alpha1 subunit. J Biol Chem 272: 18759-65. [Crossref]
- Hockerman GH, Peterson BZ, Sharp E, Tanada TN, Scheuer T, et al (1997) Construction of a high-affinity receptor site for dihydropyridine agonists and antagonists by single amino acid substitutions in a non-L-type Ca2+ channel. Proc Natl Acad Sci USA 94: 14906-14911. [Crossref]
- Striessnig J (1999) Pharmacology, structure and function of cardiac L-type Ca(2+) channels. Cell Physiol Biochem 9: 242-269. [Crossref]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, et al. (1998) Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol Sci 19: 108-115. [Crossref]
- Chen N, Zhou M, Yang M, Guo J, Zhu C, et al. (2010) Calcium channel blockers versus other classes of drugs for hypertension. Cochrane Database Syst Rev: CD003654. [Crossref]
- Boland LM, Morrill JA, Bean BP (1994) omega-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci 14: 5011-5027. [Crossref]
- Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, et al. (1992) A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron 9: 69-77. [Crossref]
- Lewis, R.J., et al. (2000) Novel omega-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J Biol Chem 275: 35335-35344. [Crossref]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, et al. (1992) P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature 355: 827-829. [Crossref]
- Newcomb R, Szoke B, Palma A, Wang G, Chen Xh, et al (1998) Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry 37: 15353-15362. [Crossref]
- Kang HW, Park JY, Jeong SW, Kim JA, Moon HJ, et al (2006) A molecular determinant of nickel inhibition in Cav3.2 T-type calcium channels. J Biol Chem 281: 4823-4830. [Crossref]
- Díaz RA, Sancho J, Serratosa J (2008) Antiepileptic drug interactions. Neurologist 14: S55-65. [Crossref]
- Chuang RS, Jaffe H, Cribbs L, Perez-Reyes E, Swartz KJ (1998) Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nature Neurosci 1: 668-674. [Crossref]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E (1999) Nickel Block of Three Cloned T-Type Calcium Channels: Low Concentrations Selectively Block alpha1H. Biophys J 77: 3034-3042. [Crossref]
- Smith EM, Sorota S, Kim HM, McKittrick BA, Nechuta TL, et al (2010) T-type calcium channel blockers: spiro-piperidine azetidines and azetidinones-optimization, design and synthesis. Bioorg Med Chem Lett 20: 4602-6. [Crossref]
- Lacinová L (2005) Voltage-dependent calcium channels. Gen Physiol Biophys 24 Suppl 1: 1-78. [Crossref]
- Missiaen L, Robberecht W, van den Bosch L, Callewaert G, Parys JB, et al. (2000) Abnormal intracellular ca(2+)homeostasis and disease. Cell Calcium 28: 1-21. [Crossref]
- Gibney GT, Zhang JH, Douglas RM, Haddad GG, Xia Y (2002) Na(+)/Ca(2+) exchanger expression in the developing rat cortex. Neuroscience 112: 65-73. [Crossref]
- Kannurpatti SS, Joshi PG, Joshi NB (2000) Calcium sequestering ability of mitochondria modulates influx of calcium through glutamate receptor channel. Neurochem Res 25: 1527-1536. [Crossref]
- Albowitz B, König P, Kuhnt U (1997) Spatiotemporal distribution of intracellular calcium transients during epileptiform activity in guinea pig hippocampal slices. J Neurophysiol 77: 491-501. [Crossref]
- Avanzini G, Franceschetti S (2003) Cellular biology of epileptogenesis. Lancet Neurol 2: 33-42. [Crossref]
- DeLorenzo RJ, Waterhouse EJ, Towne AR, Boggs JG, Ko D, et al (1998) Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 39: 833-840. [Crossref]
- Pisani A, Bonsi P, Martella G, De Persis C, Costa C, et al. (2004) Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia 45: 719-728. [Crossref]
- Beck H, Steffens R, Elger CE, Heinemann U (1998) Voltage-dependent Ca2+ currents in epilepsy. Epilepsy Res 32: 321-332. [Crossref]
- Köhling R, Straub H, Speckmann EJ (2000) Differential involvement of L-type calcium channels in epileptogenesis of rat hippocampal slices during ontogenesis. Neurobiol Dis 7: 471-82. [Crossref]
- Speckmann EJ, Straub H, Köhling R (1993) Contribution of calcium ions to the generation of epileptic activity and antiepileptic calcium antagonism. Neuropsychobiology 27: 122-126. [Crossref]
- Straub H, Speckmann EJ, Bingmann D, Walden J (1990) Paroxysmal depolarization shifts induced by bicuculline in CA3 neurons of hippocampal slices: suppression by the organic calcium antagonist verapamil. Neurosci Lett 111: 99-101. [Crossref]
- Becker AJ, Pitsch J, Sochivko D, Opitz T, Staniek M, et al. (2008) Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J Neurosci 28: 13341-13353. [Crossref]
- Chuang, R.S.-I., et al (1998) Identification of an inhibitor of T-type calcium channels. Soc Neurosci Abstr 24: 22.
- Zaman T, Lee K, Park C, Paydar A, Choi JH, et al. (2011) Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron 70: 95-108. [Crossref]
- Kuzmiski JB, Barr W, Zamponi GW, MacVicar BA (2005) Topiramate inhibits the initiation of plateau potentials in CA1 neurons by depressing R-type calcium channels. Epilepsia 46: 481-489. [Crossref]
- Tai C, Kuzmiski JB, MacVicar BA (2006) Muscarinic enhancement of R-type calcium currents in hippocampal CA1 pyramidal neurons. J Neurosci 26: 6249-6258. [Crossref]
- Wojda U, Salinska E, Kuznicki J (2008) Calcium ions in neuronal degeneration. IUBMB Life 60: 575-590. [Crossref]
- Besancon E, Guo S, Lok J, Tymianski M, Lo EH (2008) Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci,. 29: 268-75. [Crossref]
- Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, et al. (2011) L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer's disease. Neurobiol Dis 41: 62-70. [Crossref]
- Goodison WV, Frisardi V, Kehoe PG (2012) Calcium channel blockers and Alzheimer's disease: potential relevance in treatment strategies of metabolic syndrome. J Alzheimers Dis 30 Suppl 2: S269-282. [Crossref]
- Weiergräber M, Henry M, Krieger A, Kamp M, Radhakrishnan K, et al. (2006) Altered seizure susceptibility in mice lacking the Ca(v)2.3 E-type Ca2+ channel. Epilepsia 47: 839-850. [Crossref]
- Weiergräber M, Kamp MA, Radhakrishnan K, Hescheler J, Schneider T (2006) The Ca(v)2.3 voltage-gated calcium channel in epileptogenesis--shedding new light on an enigmatic channel. Neurosci Biobehav Rev 30: 1122-1144. [Crossref]
- Khosravani H, Zamponi GW (2006) Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol Rev 86: 941-966. [Crossref]
- Kim S, Rhim H (2011) Effects of amyloid-β peptides on voltage-gated L-type Ca(V)1.2 and Ca(V)1.3 Ca(2+) channels. Mol Cells 32: 289-294. [Crossref]
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, et al. (2010) Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 107: 14950-14957. [Crossref]
- Bredesen DE, Rao RV, Mehlen P (2006) Cell death in the nervous system. Nature 443: 796-802. [Crossref]
- Ben-Ari Y, Cossart R (2000) Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci 23: 580-587. [Crossref]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, et al. (1996) Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature 379: 78-81. [Crossref]
- Castillo PE, Malenka RC, Nicoll RA (1997) Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388: 182-186. [Crossref]
- Lerma J, Paternain AV, Rodríguez-Moreno A, López-García JC (2001) Molecular physiology of kainate receptors. Physiol Rev 81: 971-998. [Crossref]
- Lerma J (2003) Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci 4: 481-495. [Crossref]
- Nicoll RA, Schmitz D (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci 6: 863-876. [Crossref]
- Schmitz D, Mellor J, Frerking M, Nicoll RA (2001) Presynaptic kainate receptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A 98: 11003-11008. [Crossref]
- Schmitz D, Mellor J, Nicoll RA (2001) Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 291: 1972-1976. [Crossref]
- Contractor A, Swanson G, Heinemann SF (2001) Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron 29: 209-216. [Crossref]
- Dietrich D, Kirschstein T, Kukley M, Pereverzev A, von der Brelie C, et al. (2003) Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron 39: 483-496. [Crossref]
- Hunsberger JG, Bennett AH, Selvanayagam E, Duman RS, Newton SS (2005) Gene profiling the response to kainic acid induced seizures. Brain Res Mol Brain Res 141: 95-112. [Crossref]
- Krieger A, Radhakrishnan K, Pereverzev A, Siapich SA, Banat M, et al. (2006) The molecular chaperone hsp70 interacts with the cytosolic II-III loop of the Cav2.3 E-type voltage-gated Ca2+ channel. Cell Physiol Biochem 17: 97-110. [Crossref]
- Meza U, Thapliyal A, Bannister RA, Adams BA (2007) Neurokinin 1 receptors trigger overlapping stimulation and inhibition of Cav2.3 (R-type) calcium channels. Mol Pharmacol 71: 284-293. [Crossref]
- Yagami T, Kohma H, Yamamoto Y (2012) L-type voltage-dependent calcium channels as therapeutic targets for neurodegenerative diseases. Curr Med Chem 19: 4816-4827. [Crossref]
- Brewer LD, Thibault O, Staton J, Thibault V, Rogers JT, et al (2007) Increased vulnerability of hippocampal neurons with age in culture: temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res 1151: 20-31. [Crossref]
- Colwell CS, Levine MS (1999) Metabotropic glutamate receptor modulation of excitotoxicity in the neostriatum: role of calcium channels. Brain Res 833: 234-241. [Crossref]
- Cater HL, Gitterman D, Davis SM, Benham CD, Morrison B 3rd, et al (2007) Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. J. Neurochem 101: 434-447. [Crossref]
- Bloodgood BL, Sabatini BL (2007) Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol 17: 345-351. [Crossref]
- Bloodgood BL, Sabatini BL (2007) Nonlinear regulation of unitary synaptic signals by Cav2.3 voltage-sensitive calcium channels located in dendritic spines. Neuron 53: 249-60. [Crossref]
- Yasuda R, Sabatini BL, Svoboda K (2003) Plasticity of calcium channels in dendritic spines. Nat Neurosci 6: 948-955. [Crossref]
- Schulz R, Kirschstein T, Brehme H, Porath K, Mikkat U, et al (2012) Network excitability in a model of chronic temporal lobe epilepsy critically depends on SK channel-mediated AHP currents. Neurobiol Dis 45: 337-347. [Crossref]
- Metz AE, Jarsky T, Martina M, Spruston N (2005) R-type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci 25: 5763-5773. [Crossref]
- Weiergräber M, Henry M, Krieger A, Kamp M, Radhakrishnan K, et al. (2006) Altered seizure susceptibility in mice lacking the Ca(v)2.3 E-type Ca2+ channel. Epilepsia 47: 839-850. [Crossref]
- Weiergräber M, Kamp MA, Radhakrishnan K, Hescheler J, Schneider T (2006) The Ca(v)2.3 voltage-gated calcium channel in epileptogenesis--shedding new light on an enigmatic channel. Neurosci Biobehav Rev 30: 1122-1144. [Crossref]
- Weiergräber M, Henry M, Radhakrishnan K, Hescheler J, Schneider T (2007) Hippocampal seizure resistance and reduced neuronal excitotoxicity in mice lacking the Cav2.3 E/R-type voltage-gated calcium channel. J Neurophysiol 97: 3660-3669. [Crossref]
- Klöckner U, Pereverzev A, Leroy J, Krieger A, Vajna R, et al (2004) The cytosolic II-III loop of Cav2.3 provides an essential determinant for the phorbol ester-mediated stimulation of E-type Ca2+ channel activity. Eur J Neurosci 19: 2659-2668. [Crossref]
- Suzuki T, Delgado-Escueta AV, Aguan K, Alonso ME, Shi J, et al. (2004) Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet 36: 842-849. [Crossref]
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, et al. (2002) Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci 22: 3645-3655. [Crossref]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, et al. (2004) Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119: 19-31. [Crossref]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, et al. (2005) Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A 102: 8089-8096. [Crossref]
- Pietrobon D (2013) Calcium channels and migraine. Biochim Biophys Acta 1828: 1655-1665. [Crossref]
- Pietrobon D, Moskowitz MA (2013) Pathophysiology of migraine. Annu Rev Physiol 75: 365-391. [Crossref]
- Pietrobon D (2010) CaV2.1 channelopathies. Pflugers Arch 460: 375-393. [Crossref]
- Takeda A (2001) Zinc homeostasis and functions of zinc in the brain. Biometals 14: 343-351. [Crossref]
- Mathie A, Sutton GL, Clarke CE, Veale EL (2006) Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther 111: 567-583. [Crossref]
- Kask K, Jerecic J, Zamanillo D, Wilbertz J, Sprengel R, et al. (2000) Developmental profile of kainate receptor subunit KA1 revealed by Cre expression in YAC transgenic mice. Brain Res 876: 55-61. [Crossref]
- Ono S, Cherian MG (1999) Regional distribution of metallothionein, zinc, and copper in the brain of different strains of rats. Biol Trace Elem Res 69: 151-159. [Crossref]
- Sato M, Ohtomo K, Daimon T, Sugiyama T, Iijima K (1994) Localization of copper to afferent terminals in rat locus ceruleus, in contrast to mitochondrial copper in cerebellum. J Histochem Cytochem 42: 1585-91. [Crossref]
- Ayton S, Lei P, Bush AI (2013) Metallostasis in Alzheimer's disease. Free Radic Biol Med 62: 76-89. [Crossref]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, et al. (1996) The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272: 1013-1016. [Crossref]
- Kim YH, Kim EY, Gwag BJ, Sohn S, Koh JY (1999) Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience 89: 175-182. [Crossref]
- Sensi SL, Yin HZ, Weiss JH (2000) AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur. J. Neurosci 12: 3813-3818. [Crossref]
- Weiss JH, Sensi SL (2000) Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci 23: 365-371. [Crossref]
- Takeda A, Minami A, Seki Y, Oku N (2003) Inhibitory function of zinc against excitation of hippocampal glutamatergic neurons. Epilepsy Res 57: 169-174. [Crossref]
- Takeda A, Hirate M, Tamano H, Nisibaba D, Oku N (2003) Susceptibility to kainate-induced seizures under dietary zinc deficiency. J Neurochem 85: 1575-1580. [Crossref]
- Takeda A, Tamano H, Nagayoshi A, Yamada K, Oku N (2005) Increase in hippocampal cell death after treatment with kainate in zinc deficiency. Neurochem Int 47: 539-544. [Crossref]
- Takeda A, Yamada K, Minami A, Nagano T, Oku N (2005) Enhanced excitability of hippocampal mossy fibers and CA3 neurons under dietary zinc deficiency. Epilepsy Res 63: 77-84. [Crossref]
- Pei Y, Zhao D, Huang J, Cao L (1983) Zinc-induced seizures: a new experimental model of epilepsy. Epilepsia 24: 169-176. [Crossref]
- Fukahori M, Itoh M (1990) Effects of dietary zinc status on seizure susceptibility and hippocampal zinc content in the El (epilepsy) mouse. Brain Res 529: 16-22. [Crossref]
- Williamson A, Spencer D (1995) Zinc reduces dentate granule cell hyperexcitability in epileptic humans. Neuroreport 6: 1562-1564. [Crossref]
- Sík A, Hájos N, Gulácsi A, Mody I, Freund TF (1998) The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci U S A 95: 3245-3250. [Crossref]
- Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H (1994) Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron 12: 1281-1289. [Crossref]
- Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH (1999) Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci U S A 96: 2414-2419. [Crossref]
- Côté A, Chiasson M, Peralta MR 3rd, Lafortune K, Pellegrini L, et al. (2005) Cell type-specific action of seizure-induced intracellular zinc accumulation in the rat hippocampus. J Physiol 566: 821-837. [Crossref]
- Pisani A, Bonsi P, Bernardi G, Calabresi P (2002) Impairment of mitochondrial metabolism differentially affects striatal neuronal subtypes. Neuroreport 13: 641-644. [Crossref]
- Lavoie N, Peralta MR 3rd, Chiasson M, Lafortune K, Pellegrini L, et al (2007) Extracellular chelation of zinc does not affect hippocampal excitability and seizure-induced cell death in rats. J. Physiol 578: 275-289. [Crossref]
- Domínguez MI, Blasco-Ibáñez JM, Crespo C, Marqués-Marí AI, Martínez-Guijarro FJ (2003) Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience 116: 791-806. [Crossref]
- Sun HS, Hui K, Lee DW, Feng ZP (2007) Zn2+ sensitivity of high- and low-voltage activated calcium channels. Biophys J 93: 1175-1183. [Crossref]
- Takahashi K, Akaike N (1990) Nicergoline inhibits T-type Ca2+ channels in rat isolated hippocampal CA1 pyramidal neurones. Br J Pharmacol 100: 705-710. [Crossref]
- Traboulsie A, Chemin J, Chevalier M, Quignard JF, Nargeot J, et al. (2007) Subunit-specific modulation of T-type calcium channels by zinc. J Physiol 578: 159-171. [Crossref]
- Kerchner GA, Canzoniero LM, Yu SP, Ling C, Choi DW (2000) Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J Physiol 528: 39-52. [Crossref]
- Minami A, Sakurada N, Fuke S, Kikuchi K, Nagano T, et al. (2006) Inhibition of presynaptic activity by zinc released from mossy fiber terminals during tetanic stimulation. J Neurosci Res 83: 167-176. [Crossref]
- Breustedt J, Vogt KE, Miller RJ, Nicoll RA, Schmitz D (2003) Alpha1E-containing Ca2+ channels are involved in synaptic plasticity. Proc Natl Acad Sci U S A 100: 12450-12455. [Crossref]
- Li YV, Hough CJ, Sarvey JM (2003) Do we need zinc to think? Sci STKE 2003: pe19. [Crossref]
- Murakami K, Whiteley MK, Routtenberg A (1987) Regulation of protein kinase C activity by cooperative interaction of Zn2+ and Ca2+. J Biol Chem 262: 13902-13906. [Crossref]
- Lee JY, Park J, Kim YH, Kim DH, Kim CG, et al. (2000) Induction by synaptic zinc of heat shock protein-70 in hippocampus after kainate seizures. Exp Neurol 161: 433-441. [Crossref]
- Zamponi GW, Bourinet E, Snutch TP (1996) Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J Membr Biol 151: 77-90. [Crossref]
- Jeong SW, Park BG, Park JY, Lee JW, Lee JH (2003) Divalent metals differentially block cloned T-type calcium channels. Neuroreport 14: 1537-1540. [Crossref]
- Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, et al. (2007) Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J Neurosci 27: 8250-8260. [Crossref]
- Kang HW, Moon HJ, Joo SH, Lee JH (2007) Histidine residues in the IS3-IS4 loop are critical for nickel-sensitive inhibition of the Cav2.3 calcium channel. FEBS Lett 581: 5774-5780. [Crossref]
- Shcheglovitov A, Vitko I, Lazarenko RM, Orestes P, Todorovic SM, et al. (2012) Molecular and biophysical basis of glutamate and trace metal modulation of voltage-gated Ca(v)2.3 calcium channels. J Gen Physiol 139: 219-234. [Crossref]
- Vlachová V, Zemková H, Vyklický L Jr (1996) Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur J Neurosci 8: 2257-2264. [Crossref]
- Shcheglovitov A, Vitko I, Lazarenko RM, Orestes P, Todorovic SM, et al. (2012) Molecular and biophysical basis of glutamate and trace metal modulation of voltage-gated Ca(v)2.3 calcium channels. J Gen Physiol 139: 219-234. [Crossref]
- Stea A, Soong TW, Snutch TP (1995) Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron 15: 929-940. [Crossref]
- Stea A, Soong TW, Snutch TP (1995) Voltage-Gated Calcium Channels, in Handbook of Receptors and Channels: Ligand- and voltage-gated Jon channels, R.A. North, Editor, CRC Press: Boca Raton, Ann Arbor, London, Tokyo 113-151.
- Dawson AP, Comerford JG, Fulton DV (1986) The effect of GTP on inositol ,4,5-trisphosphate-stimulated Ca2+ efflux from a rat liver microsomal fraction. Is a GTP-dependent protein phosphorylation involved? Biochem J 234: 311-315. [Crossref]
- Dawson CM, Lebrun P, Herchuelz A, Malaisse WJ, Gonçalves AA, et al. (1986) Effect of temperature upon potassium-stimulated insulin release and calcium entry in mouse and rat islets. Horm Metab Res 18: 221-224. [Crossref]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL (1992) The time course of glutamate in the synaptic cleft. Science 258: 1498-1501. [Crossref]
- Mathie A (2007) Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J Physiol 578: 377-385. [Crossref]
- Osterberg R (1980) Physiology and pharmacology of copper. Pharmacol Ther 9: 121-146.
- Kay AR (2004) Detecting and minimizing zinc contamination in physiological solutions. BMC Physiol 4: 4. [Crossref]
- Weiergraber M, Hescheler J, Schneider T (2008) Human calcium channelopathies. Voltage-gated Ca2+ channels in etiology, pathogenesis, and pharmacotherapy of neurologic disorders. Nervenarzt 79: 426-436.