Abstract
Recent studies in cystic fibrosis (CF) transmembrane regulator (CFTR) mutations and function have shed light on its involvement in disease progression. The extent of cell and tissue distribution of CFTR facilitates systemic dysfunction of ion transport in patients carrying a mutation in CFTR, however, its incidences as co-founding risk factor to develop other diseases is not well studied. In this review we differentiate the dysfunctions driven by CFTR mutations in cell of the immune system and their role in CF progression and examine the types of medical treatments available to patients up to date.
Key words
cystic fibrosis, CFTR-deficiency, CF-immune response, antibiotic therapies, microbioma maintenance, and gene therapy
Introduction
Identification of mutations associated to Cystic Fibrosis (CF) was initially searched in lung cell, epithelia, mucus goblet cells, etc. However, recently it has become more obvious that cystic fibrosis transmembrane regulator (CFTR) mutation on immune cells produces dysregulation in the lung even before infections occur. In this review we summarized how affects immune cells response and how it has changed the treatments of CF.
Types of mutations of the cystic fibrosis transmembrane regulator
Cystic Fibrosis is an autosomal life threatening recessively inherited human disease assigned to the Mendelian Inherited disease in Man (MIM) number 219700. Clinical manifestations of the CF pathology are mainly associated to the impairing functions of gastrointestinal and respiratory tracts. Pathological characteristic of CF are commonly associated to exocrine pancreatic insufficiency, recurrent lung infections, intestinal manifestations and “salty” sweat. Also secondary pathologies such as liver disease, pancreatitis, and diabetes were observed in some patients [1].
The frequency of CF is in average 1 in 2,500-3,000 live births, meaning about 80,000 people worldwide and about 1,000 new cases of CF are diagnosed yearly. However, the probability of CF frequency is related to the geographic distribution and ethnicity. In Europe, the highest prevalence of CF is observed in Ireland (1/1,700) and the lowest in Finland (1/7,700). In the United States, the birth frequency affected of CF is 1/2,500 births for Caucasians of Northern European descent, 1/3,500 for Hispanics, 1/15,100 for Africans and 1/35,000 for Asians. The life expectancy of CF patients is increasing based on novel therapeutic strategies and novel technologies. During the 80`s the patient average life expectancy was about 14 years, increased during the 90`s to around 18 years and today is in between 30 to 40 years old [2]. However, there are huge differences between the patient’s life expectancies in developed and undeveloped countries.
The CF disorder is produced by mutations in a gene located in the human long arm of chromosome 7 (specifically 7q31.2) and assigned to abnormal CFTR protein mainly expressed in secretory epithelial cells. The CFTR is mainly located in cytosol (about 85%), meanwhile the other 15% belongs to the lumen and ER membrane. From the structural point of view, the CFTR possess three different types of domains: two membrane spanning domains, one regulator, and two nucleotide binding domains for cAMP and cGMP [3,4]. Up today, 1997 mutations have been identified in the CFTR gene reported by the Cystic Fibrosis Data Base (http://www.genet.sickkids.on.ca/CFTR/). The large amounts of CFTR mutations are related to sequence and/or promoter variation, missense and non-sense, sequence variation, frame in/del, large in/del, splicing and unidentified types involving mutations in different exons and/or introns. From physiological and therapeutic point of view, the mutations were identified roughly into six classes [5] (Table 1).
Table 1. Classification and description of cystic fibrosis mutations
Class |
Mutation |
Cellular and/or molecular phenotype |
I |
Trp1282X, and Gly542X |
Absent CFTR synthesis due to non-sense mutations, or frame shift mutations, or abnormal mRNA splicing. |
II |
Phe508del |
Defective intracellular processing of CFTR with less than normal amounts of CFTR protein at the apical plasma membrane. |
III |
Gly551Asp |
Defective regulation of CFTR channels at the apical plasma membrane. |
IV |
Arg117His |
Defective transport of anions through CFTR channels at the apical plasma membrane. |
V |
3849+10KbC>T |
Reduced production or processing of normal CFTR. |
VI |
Gln1412X |
Altered apical membrane residence time of CFTR channels with truncated c-termini. |
The types 1 to 3 are most prevalent mutations and related to pancreatic insufficiency, meanwhile types 4 to 6 are not. Particularly, class 2 is the dominant CF-mutation in Caucasians affecting 86% CF alleles. The class 2 mutation is based on the absence of three nucleotides coding for phenylalanine amino acid in the 508 position of the encoded protein named F508del [1]. Also, the frequency of F508del is variable according to the population. For example, F508del is affecting the 82% of CF patients in Denmark and 32% in Turkey [5].
CFTR function
The basis of CF pathology is linked directly to malfunction of the Cystic Fibrosis Transmembrane Regulator (CFTR) protein associated to the trans-epithelial chlorine conduction, but also implicated in the transport of other molecules through the membrane [6,7]. The physiological role of CFTR is complex because intervenes in the regulation of other channels like Renal Outer Medullary potassium channel (ROMK K1+), Epithelial sodium (Na1+) Channel (ENaC), Outwardly Rectifying Cl1- channel (ORCC), and Cl1-/HCO31- exchanger [8-10]. The CFTR protein is a channel member of the ATP-binding cassette superfamily (ABC transporters) commonly described as ATP hydrolyzing proteins able to transport different molecules through cellular membranes against concentration gradients [11]. The CFTR transporter is classified as ABCC7 found in almost any form of life but with different ion permeability [12]. Also, the CFTR transporter in humans is naturally expressed in many types of cells like lung microvasculature and epithelia, pancreas, liver, heart myocytes, brain (hypothalamus), colon. The CFTR gene sequence is over 300 Kb pairs containing 27 exons and coding for a protein of 1480 base pairs with approximately 180 kD MW containing approximately 7% of glycosylated residues (galactose, fucose and sialic acid).
CFTR structure
From structural point of view, the CFTR protein is arranged in 7 domains with two amino and carboxyl termini located in the cytoplasm, two membrane-spanning domains containing six membrane-spanning segments, a regulatory domain and two nucleotide binding domains named NBD1 and NBD2 (Figure 1).
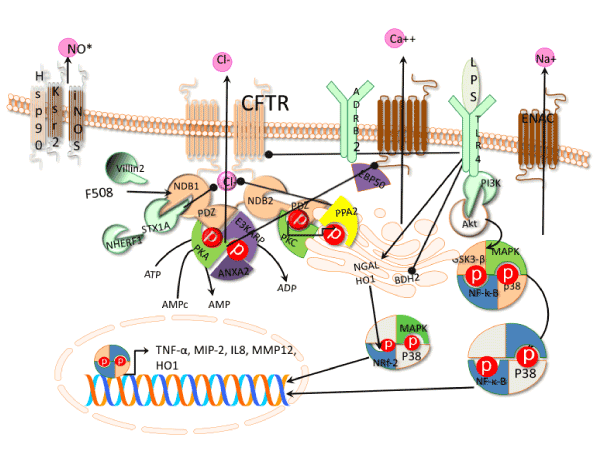
Figure 1. Signaling pathways involved in CFTR regulation. Beta-2 adrenergic receptor (ADRB2), phosphatases type II (PP2A), Protein Kinases A (PKA), subunit PRKAR2A, Protein Kinases C (PKC), mitogen activated protein kinases (MAPKs) receptor for the binding of epinephrine (ADRB2), syntaxin 1A (STX1A), Phosphatidil inositol 3 kinase (PI3K). Nuclear Factor-κ B (NF-κ B) repressor (GSK3β), nuclear factor erythroid 2–related factor 2 (Nrf2), Toll like receptor 4 (TLR4), lipopolysaccharide (LPS), macrophage inducible protein-2 (MIP-2) by p38. Macrophage inducible protein-2 (MIP-2), Tumor necrosis factor a. Macrophage metallo elastase 12 (MMP12), butyrate dehydrogenase type-2 (BDH2), iron carrier protein NGAL, heme oxygenase1 (HO1), βepithelial Na+ channel transgenic mouse (ENaC). Pointed arrows means activation and bold arrows inhibition.
The structure of the two membrane domains is composed of six helixes each represent the 25% of the protein with 19% on membrane and 4% on surface. Besides, the two membrane domains are the gateway and control of chloride flux [13]. The two cytoplasmic NBDs are the regulatory domains involving the ATP binding and hydrolysis sites that induce conformational changes and dimerization [14]. The structural changes of the NBD dimer regulate the ion fluxes trough the gateway. The NDBs domains are rich in phosphorylation sites, which resemble protein kinases [14].
Regulation of the CFTR
The mechanism of CFTR regulation is very complex (Figure 1) and involves different regulatory levels such as phosphorylation and dephosphorylation, posttranslational modifications, e.g. glycosylation, to the interaction with membrane and cytoplasmic proteins, and check point of appropriate CFTR structure. The phosphorylation/dephosphorylation mechanisms on NBD domains of the CFTR are required to activate and deactivate the chloride ion channel a cAMP-dependent manner [15]. The phosphorylation of CFTR by PKA is catalyzed by the subunit PKA-cat and regulated by the subunit PRKAR2A through the villin-2 protein (also known as Ezrin) who anchors PKA-cat. Villin-2 interacts with the NBD domain of CFTR mediated by the regulatory protein E3KARP (also named as NHERF-2) containing a PDZ binding motif. The PDZ motif of E3KARP possess high affinity for the PDZ counterpart motif located at the carboxylate terminus of NDB domain and also Annexin II protein complex, involved in the interaction to enhance the phosphorylation of CFTR [16]. On the other side, activation of PKA mediated by adenosine also induced the activity of phosphodiesterase 4D (PDE4D) catalyzing the conversion of cAMP into AMP. The AMP product activates 5‘AMP activated protein kinase (AMPK) acting a negative effector by phosphorylating the NDB of CFTR and reducing the chlorine ion transport [17]. Also, protein phosphatases type II (PP2A and PP2C) dephophorylates CFTR causing inhibition of ion transport [18]. Besides, other protein complexes like the synatosomal-associated 23 kDa protein (known as SNAP-23)-syntaxin 1A interfere sterically with the CFTR transporter reducing the chloride ion transport [19].
EBP50, another protein with PDZ binding domain, is the protein bridge between Beta-2 adrenergic receptor (ADRB2) and CFTR. However, the complex formation with the beta-2 adrenergic receptor is inhibited when the PKA phosphorylates the CFTR. The ADRB2 is a receptor for the binding of epinephrine and linked to the calcium ion channel, which is functional for smooth muscle relaxation and bronchodilation [20]. Another way to inhibit the CFTR is by suppressing the activity of PKA and adenylate cyclases depressing the cAMP in the cell by members of G-protein a-I family cascade [21].
The regulation of the CFTR function involves also the interaction of the transporter with cytoplasmic proteins like NHERF-1 and regulating the ion flux at the gateway [22]. In addition, the endoplasmatic reticulum (ER) is working as "custom" of the CFTR transporter checking proper protein folding and glycosilation. Proteolysis of abnormal CFTR transporter was suggested by tagging the protein with an "adaptor ligase" activating the ubiquitin - proteasome degradative pathway associated to ER named as ERAD [23,24].
On the other hand, CFTR regulation is not only made in the cell cytoplasm but also in the cell membrane. CFTR was phosphorylated by Protein Kinase C-e (epsilon) in order to stabilize the transporter in the membrane and also to prime the PKA. However, the PKC-e is part of the protein complex pathway involving soluble carrier family 9 (member 3 regulator 1), guanine nucleotide binding protein b-polyppetide2-like 1 [25,26].
CFTR mutations on inflammatory cells
It is thought that CF is an epithelia based disease; however CFTR dysfunction of immune cells plays a causal role in lung infection as CFTR mutations predisposes to Th2 bias that differentiates CF immune response from the rest of the lung diseases, and this precedes lung infections (Figure 2). In this review we will focus on the changes produced by CFTR mutations on immune cells [27,28].
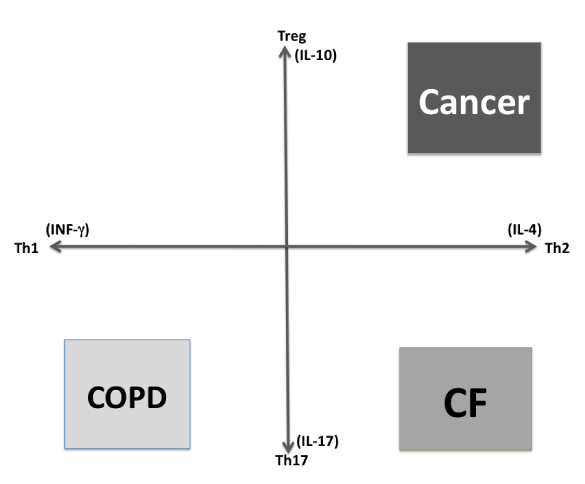
Figure 2. Schematic representation of lung immune response biases. Chronic obstructive pulmonary disease (COPD). Cystic fibrosis (CF).
Lymphocytes
In order to achieve homeostasis after an inflammatory process has been initiated in response to cytokine stimulation, T helper’s naïve (Th0) cells are differentiated along Th1/Th2 and Th17/ T regulatory (Treg) lineages, which suppress the inflammation. Recently was shown that Tregs from patients with CF, or from knock out CFTR mice (CFTR/), are impaired in suppressing conventional T effector cells, an effect that was enhanced by P. aeruginosa infection. The loss of Tregs in CF affected memory, but not naïve, Tregs and manifested gradually with disease progression, confirmed by a positive association between lung function, measured by forced expiratory volume (FEV1%), and number of Tregs present in peripheral blood of CF patients [29,30].
Murine model of CF confirmed impaired Tregs down regulation of Th2 cells; CFTR-/- mice had enhanced intracellular Ca2+ flux in response to T cell receptor (TCR) activation on T effectors cells, higher interleukin (IL) 4, IL-13, IgE and IgG1, this prominent Th2 bias correlated with higher nuclear NFAT level than their wild type control littermates (CFTR+/+). There was also an impaired inactivation of these cells by Tregs and defective Th1 response, indicating the CFTR importance to maintain the Th1/Th2 balance [30-32].
Th2 bias assessed in CF patients preceded the P. aeruginosa infection in a study conducted in 75 subjects, which proved a positive association among secretion of IL-5 with number of neutrophils in broncho-alveolar lavage (BAL) of CF subjects. There was a significant association among computed tomography changes and cytokines and chemokines assessed for Th2 bias (IL5, IL13, TARC/CCL17) and Th17 (IL17A, IL6, IL1β, IL8) [33]. However, IL-13 seems to play a greater role then IL-4 in CF since lung parenchyma leukocytes produce high IL-13, IL-10 and RANTES levels and weakly IL-17, when exposed to Pseudomonas sp.
The signaling path that determines Th2 bias includes the purinergic receptor (P2Y2) which senses ATP level secreted in a CFTR dependent manner in the first stage of inflammation upon infection (Figure 3) [34,35]. Upon viral infections knockout mice in P2Y2 had increased IL-6, reduced ATP, IL-12, and CD4+, CD8+ T cells and dendritic cell, with defective viral clearance and predominant Th2 bias but normal levels of IP10 and MIP-3 as represented in Figure 3 [35].
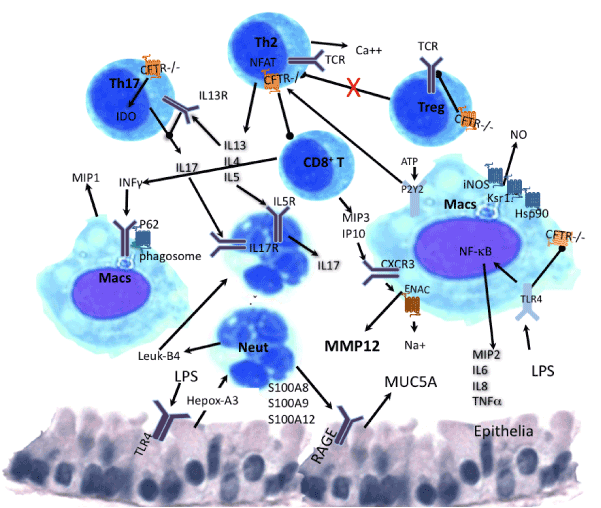
Figure 3. Schematic representation of immune cross-talk among cells CFTR deficient. Cells represented are Macrophages (Macs), T helpers 2 (Th2), T regulatory (Treg) and Th17, neutrophils (Neut), mediators are Interleukins (IL), nitric oxide (NO), interferon gamma (INFg), interferon gamma inducible protein 10 (IP10), chemokine receptor 3 (CXCR3), leukotriene B4 (leuk4), hepoxilin A3, macrophage inducible proteins (MIP) 1, 2, 3. Autophagy protein p62, Scaffold protein Ksr1, heat shock protein 90 (Hsp90), nitrogen oxide synthase (iNOS), T cell receptor (TCR), secretion proteins (S100A8, S100A9 and S100A12), receptor for advanced glycation endproducts (RAGE). Mucine 5 A (MUC5A), indole-amine 2,3dioxygenase (IDO). Th2 bias includes the purinergic receptor (P2Y2). Pointed arrows means activation and bold arrows inhibition.
In addition to Th2 bias, the binding of IL-13 to its receptors on Th17 cells down regulates IL-17A production (Figure 3) and its neutralization is necessary for significant IL-17 augmentation, this increase was a lesser amount with IFN-γ or IL-4 neutralization [27,28]. Consequently, T cells specific for Pseudomonas aeruginosa and Aspergillus fumigatus, obtained from patients with CF, displayed significantly higher IL5 and IL17A mRNA expression and TARC/CCL17 in patients that developed P. aeruginosa infection within 24 months. High expression of these cytokines/chemokines in CF airways preceded infection with P. aeruginosa demonstrating an intrinsic predisposition of Th0 cells to differentiate to Th17 phenotype although they had normal capacity for Th1 and Treg differentiation [33,36,37].
Th17, IL-17+ neutrophils, γδT and natural killer T cells (NKT) are increased in newly diagnosed CF subjects with BAL fluid neutrophilia [38]. Th17 cells from CF subjects had an antigen-specific response rather than a merely nonspecific mitogen-induced IL-17 release. Within the CD4+IL-17+ population there were some expressing IL-22 with a higher proportion of CD45RO+TCRαβ+ cells compared with IL-22- cells, suggesting a role for IL-22 in maturation of the adaptive response to develop memory T-cell fraction [27]. The mechanism of Th17 production involves indole-amine 2,3dioxygenase (IDO), since in murine and human CF dysfunctional IDO activity correlated with imbalanced Th17/Treg cell responses to Aspergillus fumigatus, that was restored by genetic recue of IDO production as represented in Figures 1 and 2 [39].
Tregs are essential to maintain immune system homeostasis by suppresing T effector cells harnessing tissue destruction; in healthy humans, fungusspecific CD4+CD25+CD127Foxp3+ Treg are strongly expanded in peripheral blood and possess phenotypic, epigenetic and functional features of thymusderived Treg. However, for Aspergillus sp., the strong Treg response contrasts with minimal conventional Tcell memory, indicating selective Treg expansion as an effective mechanism to prevent inappropriate immune activation in healthy individuals. By contrast, in subjects with Aspergillus sp. allergies, specific Th2 cells were strongly expanded despite the presence of specific Treg [40]. Upon P. aeruginosa infection CFTR-/- mice had reduced Treg count, while in CF patients there was impaired Treg suppression of Th2 inflammation, indicating the same impairment that underlay Aspergillus sp. infection [30].
It has emerged autoimmunity particularly overt in cystic fibrosis mice lacking invariant natural killer T (iNKT) cells which play a role to control autoimmune diseases triggered by a ceramidemediated induction of cell death in CF, and although suppression of the autoimmune response by iNKT cells is beneficial, IL 17+ iNKT cells attract macrophages and neutrophils to CF lungs resulting in chronic inflammation. Genetic deletion of iNKT cells in cystic fibrosis mice prevents inflammation in their lungs [41].
In summary, CFTR deficiency is highly disruptive of T cells function promoting an exaggerated Th2 response, reduced Treg population and enhanced Th17 response, this increases neutrophils recruited by IL-17 to the lung, which are a major cause of alveoli destruction associated with lung decay in CF.
Macrophages
Cystic fibrosis patients are susceptible to infections of opportunistic extracellular or intracellular pathogens, the appropriate immune response is different for each type of infection and the capacity to resolve it favorably depends on the CFTR impairment of the immune cell type involved. CF patients with mucoid P. aeruginosa chronic infections have a positive correlation between lung function and interferon IFN-γ produced by CFTR-/- peripheral blood mononuclear cells. In contrast, IL17A levels tended to correlate negatively with lung function [42]. IFNγ induces phagocytosis binding autophagy protein p62, which promotes the double membrane vacuolization around the infecting bacteria increasing traffick to the lysosomes (Assani et al., 2014). IFNγ also up regulates monocytes the chemo-attractant protein1 (MIP-1), tumor necrosis factor α (TNFα) and IL6 production and down regulates IL10 expression in infected macrophages [42].
Macrophages with CFTR depletion from membrane lipids raft, caused by second hand smoke, lypopolysaccharide (LPS) or alginate produced by P. aeruginosa, or mutations, have inhibited their phagocytic capacity [44,45]. CFTR-/- macrophages exhibit aberrant acidification of phagolysosomes and do not kill internalized microorganisms as effectively as wild-type cells do [46].
Bacterial LPS produces uncontrolled toll like receptor 4 (TLR4) signals through phosphatidil inositol 3 kinase (PI3K), activating Akt, which inactivates the Nuclear Factor-κ B (NF-κ B) repressor (GSK3β) inducing nuclear transcriptions factor (NF-κ B), p38 and mitogen activated protein kinases (MAPKs) activity transcribing TNF-α, IL-6,IL-8 and macrophage inducible protein-2 (MIP-2) and repressing of scaffold protein CAV1. CFTR-/- macrophages have upregulated phospholipase A2α (PLA2α) activity and IL-6 secretion, which is lethal upon P. aeruginosa infection. IL-6 was decreased in PLA2α-/- mice without affecting IL-1β, or TNF-α [47-49].
The uncontrolled TLR4 signaling is due to high microRNA199a5p levels, which are PI3K/AKTdependent, However, PI3K/Akt activation does not regulate phagocytosis; rather AKT/miR199a5p/CAV1 pathway promotes migration of monocytes and neutrophils responsible of hyperinflammation, tissue damage and lung decay [49,50].
LPS triggers inflammatory response in the reticulum (Figure 1) down regulating the short chain butyrate dehydrogenase type-2 protein and up-regulating the expression of iron carrier protein NGAL and stressinducible heme degrading enzyme heme oxygenase1 (HO1) leading to iron liberation from macrophages [51]. LPS and INF-γ improves macrophages killing of P. aeruginosa inducing scaffold protein Ksr1 (a ceramide-activated, serine-threonine kinase) that recruits heat shock protein 90 (Hsp90) and nitrogen oxide (NO) synthase (iNOS) to form a membrane complex that produces NO and nitric peroxide essential for bacterial clearance, as genetic deletion of Ksr1 impairs bacterial clearing and causes mouse lethality to P. aeruginosa infection [52-54].
Mycobacterium abscessus is an intracellular macrophage pathogen that has emerged as a risk factor for CF patients. M. abscessus infection is accompanied by increased HO1 precursor of billirubin and billiverdin regulators of reactive oxygen species (ROS). The HO-1 is induced by p38 MAPKdependent, as p38 inhibitors suppressed HO1 and inhibited M. abscessus growth inside macrophages. Furthermore, inhibiting HO1 using HO1 siRNA or the HO1 upstream signaling molecule, nuclear factor erythroid 2–related factor 2 (Nrf2), with siRNA results in similar inhibition of M. abscessus [55]. Nrf2 regulates a cytoprotective response to counteract oxidative stress–induced essential for clearance of intracellular pathogens like M. abscessus but a fine equilibrium has to be maintained since decrease in Nrf2 signaling is associated with the progression of diseases driven by oxidative stress response. This indicates that targeting Nrf2 can improve innate antibacterial defenses, and restore corticosteroid responses in chronic lung diseases [56].
Viral infection of mice heterozygous in CFTR mutation (CFTR +/-) showed that TGFβdependent production of IL6 by macrophages late in infection prevents acute lung injury from influenza A virus infection. This effect in CFTR+/- mice was associated with post-infection production of TGFβ with higher IL6 and alveolar macrophages at 6 days postinfection that did not occur in Wt mice. Neutralization of β-TGF reduced IL-6 secretion but IL6 neutralization did not affect BAL macrophages and neutrophils numbers, rather it attenuated the CXCL1/KC response in both strains, and reduced IFNγ production in WT mice. All this is indicating that IL-6 is involved in activation of macrophages rather than recruitment, and the IFNγ/IP10/MMP12 pathway is essential for acute lung injury as recently the relevance of MMP12 in lung injury of CF patients was assessed with the bepithelial Na1+ channel transgenic mouse (βENaCTg) that presents dehydrated airway and increased secretion of activated MMP12 (Figure 3). They also showed elevated MMP12 activity on the surface of airway macrophages in BAL from βENaCTg mice and patients with CF. Further, they report MMP12 polymorphism associated with severity of CF, indicating a new mechanism of emphysema triggered by ionic dysregulation rather than cigarette smoke exposure [57-59].
In summary, this data suggest that macrophages may be driving lung decay in CF through activation of MMP12.
Neutrophils
Patients with CF display exaggerated neutrophilic responses to pathogens and their neutrophils have an impaired capacity to undergo apoptosis, even prior to migration to the lung, as shown for Pseudomonas colonized F508 homozygote CF blood neutrophils. The blood inflammatory markers interleukin1beta (IL1β), Creactive protein (CRP) and myeloperoxidase (MPO), are elevated during recruitment of neutrophils to the lung [60].
Neutrophils roll in the blood stream and adhere to the vasculature of the injured site where the cytokines and chemokine are being produced, after chemotaxis they egress to the lung epithelia. Neutrophils carryng CFTR mutation G551D have significantly higher expression of active adhesion molecule CD11b and of CD63 on monocytes, ex vivo exposure to ivacaftor of blood cells from G551D, but not F508del and healthy subjects, resulted in changes in CXCR2 and CD16 expression on neutrophils [61].
Neutrophils recruitment to the lung is dependent of IL-17, CXCL1/KC and IL-1 production [33,62]. Epithelia cells that have been injured signal through hepoxilin A3, a chemotactic eicosanoid that mediate the transepithelial passage of neutrophils, in response to infection (Figure 3). Neutrophilderived leukotriene B4 signaling is generated by migrated neutrophils augmenting the initial hepoxilin A3 and collectively coordinate an effective neutrophilic transepithelial migratory response [63].
Decreased IL17 and the homolog to IL1 (IL8 in mice) results from the deficiency of calciumactivated chloride channel regulators (CLCA3-/-) with reduced neutrophilic infiltration into the bronchoalveolar space during bacterial infection, compared to wildtype mice [64]. Glycosaminoglycans influence the chemokine profile of the CF lung by binding IL-8 and protecting it from proteolytic degradation. IL-8 is maintained in an active state by this glycan interaction increasing neutrophil infiltration into the lungs [65]. IL-8 recruits and activates neutrophils through G-couple chemokine receptor (CXCR1), this is regulated through elastase cleavage of the receptor mediated by PAR-2, which co-localizes with CXCR1.
Neutrophils egress from the microvasculature is mediated by VAP1/SSAO, an endothelial bound adhesion molecule with amine oxidase activity. Adherence is induced by CXCL1/KC and inhibition of VAP1/SSAO also dampenes migration of neutrophils to the lungs in response to LPS [66]. Upon arrival to the lung neutrophils interact with epithelial cells through secretion of proteins S100A8, S100A9 and S100A12 which binds TLR4 and the receptor for advanced glycation endproducts (RAGE) stimulating mucin production from epithelial cells (Figure 3) [67]. C reactive protein (CRP) generated in inflamed tissues inhibits neutrophil apoptosis via a mechanism involving activation of FccRIII (CD16) and PI3K. Inhibitor of PI3K, LY294002, increased CF neutrophils apoptosis in 50%, similar to normal patients’ neutrophil apoptosis [68]. In addition to prolonged life span, that impairs inflammation resolution, neutrophils carrying CFTR mutations have significantly higher expression of active CD11b and compromised phagocytosis, which predispose CF lungs to infection [61,69].
In the tissue, neutrophils arrest P. aeruginosa growth by chemotactic effect undergoing a respiratory burst following C3b and Fc receptor activation with extracellular release of ROS such as O2-, H2O2 and NO, consuming large amounts of O2 forcing the bacteria to enter into anaerobic metabolism [70-72]. In addition, hypochlorus acid produced by this mechanism is reduced by MPO oxidizing glutathione to glutathione sulfamides [73].
CF blood neutrophils release less granule containing MPO, neutrophil elastase (NE) and extracellular DNA compared with control patients and this is associated with reduced activity of the low molecular weight GTP-binding protein Rab27a due to higher intracellular Cl-, lower Mg2+ and reduced degranulation and killing ability [74]. Degranulation of neutrophils by gram-negative bacteria is mediated by galectin-9 (Gal9) interaction with T cell immunoglobulin and mucin domaincontaining molecule 3 (TIM-3). TIM3/Gal9 signaling modulates cytosolic Ca2+ levels and enhaces NADPH activity that is essential for ROS production [75].
Over all, the reduced apoptosis, degranulation and killing ability caused by the ionic imbalance of CFTR mutation together with increased adhesiveness leads to neutrophilia in CF lungs (Figure 3) that negatively correlates with their FEV1 [60].
CF Infections
Colonization by bacteria may synergize, amplify, or trigger pathways of tissue damage; the most common infection in cystic fibrosis patient is P. aeruginosa. However, many other opportunistic pathogens are also able to colonize the lung of these patients, infections that ultimately leads to respiratory failure.
P. aeruginosa infection
The growth rate of P. aeruginosa within CF lungs inversely correlates to the concentration of neutrophils surrounding the bacteria. A growth limiting effect on P. aeruginosa by neutrophils was also observed in vitro, where this limitation was alleviated in the presence of the alternative electron acceptor nitrate. The finding that P. aeruginosa growth patterns correlate with the number of surrounding neutrophils points to a bacteriostatic effect by neutrophils via their strong O2 consumption and release of ROS upon activation, which slows the growth of P. aeruginosa in infected CF lungs [72]. Never the less P. aeruginosa has the ability to circumvent this bactericidal mechanism producing kynurenine an oxygen peroxide and superoxide scavenger that has no effect on phagocytosis or oxygen consumption by activated neutrophils but rather in their phagosome [76].
Burkholderia cenocepacia infections
Burkholderia cenocepacia infects patients with CF within their macrophages in membrane vacuoles that preclude fusion with the lysosome [77-80]. B. cepacia complex co-infects with P. aeruginosa reducing its biofilm capacity. B. cenocepacia and B. multivorans express mucinases that utilize mucin as carbon source and fermentation rather than anaerobic respiration to obtain energy [81]. Burkholderia pseudomallei is the causative agent of melioidosis characterized by pneumonia and fatal septicemia and a strong risk factors of mortality in CF [82].
Mycobacterium abscessus infections
Mycobacterium abscessus is a rapidly growing mycobacterial species that infects macrophages, and is an important pathogen in patients with CF [55]. Genome analysis of this bacterium showed its metabolic pathways typically found in environmental microorganisms that come into contact with soil, plants, and aquatic environments but also contains several genes that are characteristically found only in pathogenic bacteria. One of them is MAB_0555, encoding a putative phospholipase C (PLC) that is absent from most other rapidly growing mycobacteria, including Mycobacterium chelonae and Mycobacterium smegmatis. PLC is highly cytotoxic to mouse macrophages, presumably due to hydrolysis of membrane phospholipids. Using an M. abscessus PLC knockout mutant was shown that loss of PLC activity is deleterious to M. abscessus intracellular survival in amoebae [83]. This ability to survive intracelullary in macrophages and neutrophils initiate the formation of cords that when released from apoptotic macrophages or neutrophil become too large to be phagocytized. Cords initiated in the vasculature enter the central nervous system producing abscesses in the brain of CF patients [84]. Aspergillus spp. are present in the airways of 2040% of CF patients and are of unclear clinical significance [31].
In addition to lung infections, patients with CF has reduced intestinal richness with decreased levels of Eubacterium rectale, Bacteroides uniformis, Bacteroides vulgatus, Bifidobacterium adolescentis, Bifidobacterium catenulatum, and Faecalibacterium prausnitzii and increased levels of inflammation shown by higher fecal levels of calprotectin and rectal nitric oxide [85]. Stimulating the growth of species involved in immunity development, such as Bifidobacterium species, Sutturella wadsworthia, and Clostridium cluster XIVa organisms, and at the same time increased the production of butyrate and propionate seems to be possible to restore Th1 bias and INF-γ levels that improves bacterial clearance [86].
Treatments of CF disease
Antibiotics resistance of pathogens infecting CF patients is a hallmark of the disease. The use of phytochemicals to enhance bacterial susceptibility to antibiotic, like zingerone (active components from ginger) seem to increase the P. aeruginosa sensitivity to antibiotics by reducing its cell surface hydrophobicity, alginate and LPS production, thus increasing its phagocytic susceptibility by macrophages decreasing TNF-a and MIP-2 produced by them [87].
In addition, alterations of the intestinal microbiota impacts the systemic immune response, reports suggest that dietary acidic oligosaccharides can favor a Th1 immune response, restoring INF-α level in the lung and bacterial clearance [86].
Antibiotics therapy
The chronic bacterial infection found in the airways is a recurrent problem in the CF disease, particularly susceptible to Pseudomonas aeruginosa, which is by far the most significant pathogen responsible of morbidity and mortality [88]. Repetitive and prolonged infections allow the selection of hyper mutated strains of P. aeruginosa with antibiotic multiresistance to conventional therapies and able to persist for years in most patients [89]. Once the strain converts to mucoid phenotype, an exacerbation of alginate secretion is produced and thereby the strain becomes extremely difficult to eradicate. In the biofilm architecture, the microorganisms are capable to survive even under aggressive antibiotic treatments [90]. Thus, an early “antibiotic eradication therapy” is highly recommended in CF [91,92]. Undoubtedly, appropriate therapies and strict use of multi-antibiotic doses are responsible of life-quality improvement and increased survival of CF patients.
Nowadays, there is not a consensus among clinicians about the most adequate antibiotic treatment against P. aeruginosa [93]. Table 2 summarizes the antimicrobial agents commonly use and their recommended doses [93]. Many factors are influencing the efficiency of drug therapies but not only based on the antibiotic therapeutic properties but also considering other factors like molecular properties of drugs (e.g. chemical structure, functional active groups, solubility, biodisponibility, target sites), the route of administration and importantly the formulation of therapeutic molecules.
Table 2. Antibiotics dosages commonly used in the management of cystic fibrosis patients with Pseudomonas
aeruginosa lung infections (modified from Döring et al., 2000).
Family |
Antibiotic |
Administration route |
Number of administrations per day |
Dose per day (mg/kg) |
Aminoglycosides |
Amikacin |
i.v. |
2 |
30 |
|
Tobramycin |
i.v. |
2 |
10 |
|
|
inhaled |
1-2 |
150-300 |
|
Netilmicin |
i.v. |
2 |
10 |
Betalactamics |
Aztreonam |
i.v. |
4 |
150 |
|
|
i.v. |
continuously |
100 |
|
Imipenem/cilastatin |
i.v. |
3-4 |
50-100 |
Cephalosporins |
Cefepime |
i.v. |
2-3 |
100-150 |
|
Ceftazidime |
i.v. |
3-4 |
150-250 |
|
|
i.v. |
continuously |
100-150 |
Quinolones |
Ciprofloxacin |
oral |
2-3 |
30 |
Polymyxins |
Colistin |
inhaled |
1-2 |
80-160 |
Carboxypenicillins |
Ticarcillin |
i.v. |
4 |
500-750 |
Carbapenems |
Meropenem |
i.v. |
3 |
60-120 |
|
|
i.v. |
continuously |
60 |
i.v. = intravenous |
Some microbial isolates from CF sputum were completely susceptible to antibiotics like: tobramycin, ceftazidime, colistin, imipenem, levofloxacin, and ciprofloxacin, and synergetic effects have been found with combination some of them [94]. The administration of inhaled antibiotics in addition to other antibiotics orally deliver in treatment for 3 weeks to 3 months has been used successfully [95]. A decrease in lung inflammation has been found in trial therapy with oral/inhaled antibiotic treatment in comparison with intravenous administration [96]. In fact, 3 months treatment with of high-doses of inhaled colistin combined with oral ciprofloxacin had produced the best therapeutic results in management of Pseudomonas aeruginosa infections [97].
Nano particles therapies
However, novel therapeutic alternatives are under intensive studies to increase the effectiveness of conventional antibiotics and to overcome the resistant mechanism of microbial pathogens. In this sense, novel strategies are focus on the antibiotic encapsulation in nano- and micro-carriers to improve the antibiotic biodisponibility, and to provide a proper molecular control release profile under the therapeutic window, avoiding the use of high concentrations which results in undesirable hepatic- and nephro-toxicity side effects. Among them, quinolones and amikacin have been encapsulated in liposomal formulation or solid lipid nanoparticles for pulmonary delivery, improving the biodistribution of the antibiotics and the patients' compliance [98]. Figure 4 shows images of amikacin loaded nanoparticles biodistribution in rats. However, the dynamic characteristics of liposomes and their intrinsic instability under different physicochemical environments do not allow predicting and determining the precise amount of delivered drug and their efficiency in the lungs. On the contrary, biopolymers are good alternative to make matrices for drug delivery since most of them are non-toxic, biodegradable, easily tailorable, and cheap. Also, biopolymer devices for drug delivery are advantageous since there are able make stable gel structures, can carry more than one therapeutic molecule, the amount of cargo/s can be very well-establish allowing to determine specific molecular kinetic release. For example, co-administration of quinolones and enzymes with complementary functions (such as alginate lyase or Dnase) encapsulated into biopolymer microspheres has been reported successful to decrease viscoelasticity of the purulent mucus and enhance the antibiotic efficiency [99,100]. The main advantage of combining antibiotics and hydrolytic enzymes are to increase not only the diffusivity of the antibiotic through the biofilms reaching the microbial cell surface more easily but also reducing the antibiotic dose. Also, the use of phytochemicals seems to enhance bacterial susceptibility to antibiotic and improve antibiofilm efficacy of ciprofloxacin against P. aeruginosa PAO1. Active components like zingerone (extracted from ginger) could increase the P. aeruginosa sensitivity to antibiotics by reducing its cell surface hydrophobicity and decreasing the alginate and LPS production. Consequently, there is an increase in its susceptibility to phagocytosis by macrophages decreasing the TNF-a and MIP-2 levels [86]. Other works reveal that extracts of garlic could act as anti-quorum sensing molecules, preventing biofilm formation, increasing the clearing of the infecting bacteria and potentiating the effect of tobramycin [101].
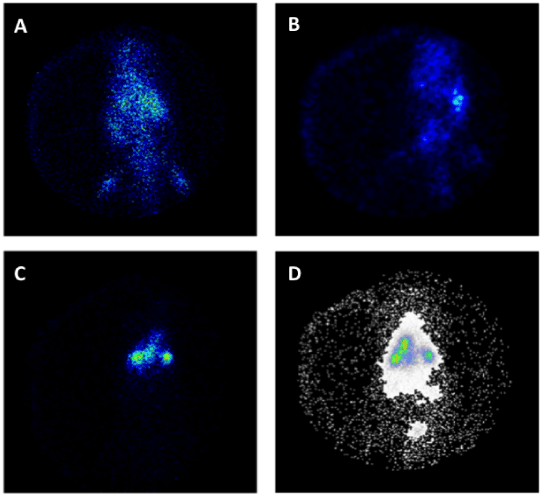
Figure 4. Gamma scintigraphy photographs of rats receiving amikacin loaded solid lipid nanoparticles (SLNs) (a) i.v. after 0.5 hour, (b) i.v. after 6 hours, (c) pulmonary after 0.5 hour, and (d) pulmonary after 6 hours (Varshosaz et al., 2013).
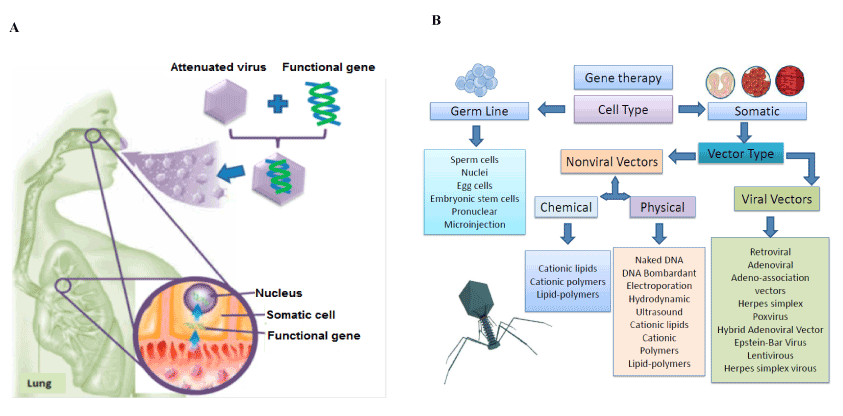
Figure 5. Scheme of the gene delivery by adenovirus to restore the defective gene a) Classification of different gene delivery systems according to the target cell (Nayerossadat et al., 2012) b) A scheme of the gene delivery by adenovirus
Microbiome maintenance
A relatively new research field is focus on the study of the lung and gastrointestinal microbiome of CF patients. Both microbiotas seem to be intrinsically influenced in such a way that the development and maintenance of lung immunity and inflammation are related with the intestinal microorganisms [102]. It was observed a strong correlation between low species richness and poor lung function [103]. Studies revealed that microorganisms in the intestine will be playing an important function in regulating T helper cells, regulatory T cells and dendritic cells as well as Toll-like receptor expression in macrophage and dendritic cells, which are the key tools to combat bacterial infections [104,105].
Patients with CF have reduced intestinal richness with low levels of Eubacterium rectale, Bacteroides uniformis, Bacteroides vulgatus, Bifidobacterium adolescentis, Bifidobacterium catenulatum, and Faecalibacterium prausnitzii, and usually present a serious degree of inflammation which is reflected in the higher fecal levels of calprotectin and rectal nitric oxide [85].
An alternative to restore the balance of the microbiome is set by stimulation of the growth of species involved in immunity development, such as Bifidobacterium and Lactobacillus species, Sutturella wadsworthia, and Clostridium cluster XIVa organisms. Studies have demonstrated that prebiotics could stimulate the growth of bifidobacteria and lactobacilli, developing important immune-modulating functions in Th1 response and T cell regulation. Short chain galacto-oligosaccharides and long-chain fructo-oligosaccharides are included in these non-digestible carbohydrates group. At the same time, prebiotics can be degraded by the natural intestinal flora to produce short-chain fatty acids (such as butyrate and propionate) which could reactivate the Th1 bias by increasing TNF-a levels that improves bacterial clearance [106].
Studies in mice models indicate that pretreatment with pectin-derived acidic oligosaccharides may help to improve the incidence of P. aeruginosa pulmonary infection by increasing bacterial clearance with a shift in the Th1/Th2 balance toward a predominantly Th1 response [86].
Certainly, further studies will be necessary to explore the microbiome of different microbial communities in the airways and intestine of healthy and infected patients to elucidate the mechanisms involved in immunity and the relationship with strong antibiotics treatments [103].
Dietary supplements
Patients with CF need to follow a very strict diet according to the age and grade of illness, and therefore different nutrient requirements should be carefully monitored to guarantee a normal growth [107]. Due to the systemic mucus accumulated at different organs, clinical compliance is associated with malnutrition and poor absorption of foods in patients [108].
The Cystic Fibrosis Foundation highlights that patients treatment include the supplying of adequate calories, intake of sufficient vitamin D and K and specially incorporation of calcium enough to avoid pathologies like osteopenia and osteoporosis [109]. Table 3 shows the most relevant nutrients that patients need to take into account for a suitable nutrition.
Table 3. Nutrients to be monitored for establish the nutritional status of CF patients (modified from Borowitz et al., 2002).
Nutrient |
Frequency to check nutrients levels |
At diagnosis |
Annual |
Other |
Vitamin A |
x |
x |
- |
Vitamin D |
x |
x |
- |
Vitamin E |
x |
x |
- |
Vitamin K |
x |
- |
Hemoptysis or hematemesis cases; liver disease |
Essential fatty acids |
- |
- |
Infants |
Calcium |
- |
- |
Patients older than 8 years (risk cases) |
Iron |
x |
x |
Poor appetite and fatigue |
Zinc |
- |
- |
Recommended 6 months supplementation and check changes in growth |
Sodium |
- |
- |
Patients exposed to heat stress or dehydration |
Protein stores |
X |
x |
Nutritional failure |
Beta carotene |
- |
- |
Under criteria of the physician |
One of the main causes of malnutrition in patients is due to a pancreatic insufficiency (PI). When PI is present, an active nutritional management is started by the addition of pancreatic enzymes to the diet. Preferably, enzymes are encapsulated in microspheres or microtablets, which provide them protection against the enteric acidity and prevent their rapid inactivation [107]. However, the enzymatic activities released in the intestine are not very efficient as it should be required by the pathology. A complementary strategy is based on the ingestion of prebiotics has a positive impact in the restoration of a health microbiota at intestine and an enhancement of the systemic immune response is usually observed. Reports suggest that dietary acidic oligosaccharides can favor a Th1 immune response, restoring INF-a level in the lung and bacterial clearance [86].
Gene transfer
The main focus for the next generation of therapeutics is based on the concepts of metabolic engineering by reprogramming cell and tissue metabolism and correcting metabolic deficiencies by changes in the somatic cell genomes using gene delivery technologies. In the last decades, different attempts have been focus in the restoration of the defective human cystic fibrosis transmembrane conductance regulator. Some experiments in transgenic mice have shown that the CFTR gen can be effectively transferred to epithelial cells of the lung. In addition, the mRNA of the gen could be found in bronchiolar and alveolar epithelial cells, while the CFTR protein in respiratory epithelial cells [110]. Similar works reported the posibility to directly transfer the normal CFTR gene to the airway epithelium by the use of a replication-deficient recombinant adenovirus vector containing the normal CFTR cDNA obtained interesting results in-vitro and in-vivo models [111]. After treatment, the absence of viral replication or virus-associated adverse effects opened a new window in the recovery of the CF lung function by gene therapy [112]. Figure 5 represents a scheme of the gene delivery by adenovirus.
In the following years, other methods for gene delivery have been explored such as fat capsules, synthetic vectors, nose drops or drizzling cells in addition to new aerosol methods using nebulizers. In this sense, novel strategies for gene therapy in CF disease are based on the discovery of more functional, effective and versatile gene delivery systems [113]. However, there is no a specific report of a gene delivery system that works properly for all kinds of cell types with no limitations or side effects. Some reviews describe in detail the different gene delivery systems for germline (like nuclei, egg cells, embryonic stem cells, pronuclear, microinjection and sperm cells) and somatic cells by the use of:
- Viral systems: such as retroviral, adenoviral, adeno-associated vectors, helper-dependent adenoviral systems, hybrid adenoviral systems, herpes simplex, pox virus, lentivirus, Epstein–Barr virus.
- Non-viral DNA based systems:
- Physical: naked DNA, DNA bombardant, electroporation, hydrodynamic, ultrasound, magnetofection.
- Chemical: cationic lipids, different cationic polymers, lipid polymers.
The selection of the adequate carrier will be restricted to the cell type, the target organ and the treatment duration [114,115]. Retroviral vectors are advantageous since they are integrating the selected recombinant genes by transduction. Particularly, lentivirus are preferred because of they are very stable vectors and able to transfect dividing and non-dividing cells by integrating the virus shell directly to the nucleus membrane of the cells. Besides, the main problem of lentivirus vectors are associated to the lack of surface proteins in the capsid for the recognition of respiratory ephitelium receptors [116], Also quiescent cells are refractory to the vector because of G0 state in where the reverse transcription is blocked [117].
Alternatively, Sendai virus (SeV) originated from mouse parainfluenza virus type 1, were explored as potential therapeutic vectors. Among the advantages of Sendai vector is a cytoplasm RNA expression without the requirement of chromosomal integration, and also displaying high transfection efficiency in epithelial cells due to the sialic acid and cholesterol receptors [118].
Among the viral vector systems, adenoviruses were the first choice of candidates for CF gene therapy since the target is the respiratory epithelium. However, adenoviruses are strong inducers of immune response and also low transduction efficiency which are limiting the therapeutic use Adeno-associated viral are an alternative vectors because of extensive gene transfer and expression in non-human epithelia and lack of strong immune response Besides, the main limitation of Adeno-associated vectors is related to the large size of the CFTR cassette to be inserted in viral capsid. Other viral vectors were reported for cystic fibrosis therapy but most of them are unstable showing inactive recombinant forms, or very low infectivity.
Overall, the most viral vector systems are made to transfect CFTR-deficient cells which require repetitive administrations and causing both cellular and humoral responses concomitantly with the decrease of gene expression However, the main concern about gene therapy is related to the safety. Viral vectors, particularly retrovirus, can elicit from mild acute inflammation to strong immune response that can compromise life. In addition, recombination events of the vector with potential wild-type viruses present in the patient can trigger unpredictable consequences
Regarding to the non-viral gene therapies, up today the transfection strategies are very limited with poor efficiencies.
Conclusions
The cystic fibrosis transmembrane regulator mutations comprise a complex pathology involving many signaling pathways and organs. Present therapies depend of case and they are determined not only by the mutations but also physiological and nutritional conditions of the patients. Up today, the therapeutic procedures are palliatives trying to keep standard levels of life quality based on combination of strict medication and nutritional diets. Nanotechnology in these aspects can provide novel platforms to treat and prevent the physiological consequences of the CF pathology. Particularly, if the pathology can be diagnosed in the patients during the early stages and the gene therapy proceeds by restoring CFTR activity in immune cells can prevent the lung pathology as well as other diseases before overlooked, like diabetes, where the role of CFTR mutations also has a role. Gene delivery seems to be a strategic area to be considered for the development of novel CF therapeutic agents based on the recent contributions of biotechnology and nanotechnologies.
References
- Lubamba B, Dhooghe B, Noel S, Leal T (2012) Cystic fibrosis: insight into CFTR pathophysiology and pharmacotherapy. Clin Biochem 45: 1132-1144. [Crossref]
- Anonymous (2015) Understanding cystic fibrosis. American Lung Association.
- Kleizen B, van Vlijmen T, de Jonge HR, Braakman I (2005) Folding of CFTR is predominantly cotranslational. Molecular Cell 20: 277-287.
- Riordan JR (2005) Assembly of functional CFTR chloride channels. Annu. Rev. Physiol. 67: 701-718.
- Ratjen F, Döring G (2003) Cystic fibrosis. Lancet 361: 681-689. [Crossref]
- Quinton PM (2008) Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet 372: 415-417. [Crossref]
- Cant N, Pollock N, Ford RC (2014) CFTR structure and cystic fibrosis. Int J Biochem Cell Biol 52: 15-25. [Crossref]
- Butterworth MB (2010) Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim Biophys Acta 1802: 1166-1177. [Crossref]
- Egan ME, Schwiebert EM, Guggino WB (1995) Differential expression of ORCC and CFTR induced by low temperature in CF airway epithelial cells. Am J Physiol 268: C243-251. [Crossref]
- Welling PA, Ho K (2009) A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297: F849-863. [Crossref]
- Klein I, Sarkadi B, Váradi A (1999) An inventory of the human ABC proteins. Biochim Biophys Acta 1461: 237-262. [Crossref]
- Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8: 67-113. [Crossref]
- Linsdell P (2006) Mechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. Exp Physiol 91: 123-129.
- Vergani P, Lockless SW, Nairn AC, Gadsby DC (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433: 876-880.
- Guggino WB, Stanton BA (2006) New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 7: 426-436. [Crossref]
- Borthwick LA, McGaw J, Conner G, Taylor CJ, Gerke V, et al. (2007) The Formation of the cAMP/Protein Kinase A-dependent Annexin 2 S100A10 Complex with Cystic Fibrosis Conductance Regulator Protein (CFTR) Regulates CFTR Channel Function. Molecular Biology of the Cell 18: 3388-3397.
- Hallows KR, McCane JE, Kemp BE, Witters LA, Foskett JK (2003) Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. The Journal of Biological Chemistry 278: 998-1004.
- Luo J, Pato MD, Rio2021 Copyright OAT. All rights reservial regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am J Physiol 274: C1397-1410. [Crossref]
- Chang SY, Di A, Naren AP, Palfrey HC, Kirk KL, et al. (2002) Mechanisms of CFTR regulation by syntaxin 1A and PKA. J Cell Sci 115: 783-791. [Crossref]
- Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, et al. (2007) Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131: 940-951. [Crossref]
- Li C, Dandridge KS, Di A, Marrs KL, Harris EL, et al. (2005) Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. The Journal of Experimental Medicine 202: 975-986.
- Raghuram V, Mak DO, Foskett JK (2001) Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ domain- mediated interaction. Proc Natl Acad Sci USA 98: 1300-1305.
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3: 100-105.
- Lederkremer GZ (2009) Glycoprotein folding, quality control and ER-associated degradation. Current Opinion in Structural Biology 19:515-523.
- Liedtke CM, Yun CH, Kyle N, Wang D (2002) Protein kinase C epsilon-dependent regulation of cystic fibrosis transmembrane regulator involves binding to a receptor for activated C kinase (RACK1) and RACK1 binding to Na+/H+ exchange regulatory factor. The Journal of Biological Chemistry 277: 22925-22933.
- Auerbach M, Liedtke CM (2007) Role of the scaffold protein RACK1 in apical expression of CFTR. Am J Physiol Cell Physiol 293: C294-304. [Crossref]
- Chan YR, K Chen, SR Duncan, KL Lathrop, JD Latoche, et al. (2013) Patients with cystic fibrosis have inducible IL-17+IL-22+ memory cells in lung draining lymph nodes. J Allergy Clin Immunol. 131: 1117-29.
- Newcomb DC, Boswell MG, Zhou W, Huckabee MM, Goleniewska K, et al. (2011) Human TH17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J Allergy Clin Immunol 127: 1006-1013. [Crossref]
- Anil N, Singh M (2014) CD4(+)CD25(high) FOXP3(+) regulatory T cells correlate with FEV1 in North Indian children with cystic fibrosis. Immunol Invest 43: 535-543. [Crossref]
- Hector A, Scha¨fer H, Po¨schel S, Fischer A, Fritzsching B, et al. (2015) Regulatory Tcell impairment in cystic fibrosis patients with chronic pseudomonas infection. Am J Respir Crit Care Med. 191: 91423.
- Allard JB, Poynter ME, Marr KA, Cohn L, Rincon M, et al. (2006) Aspergillus fumigatus generates an enhanced Th2biased immune response in mice with defective cystic fibrosis transmembrane conductance regulator. J Immunol 177: 518694.
- Mueller C, Braag SA, Keeler A, Hodges C, Drumm M, Flotte TR (2011) Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am J Respir Cell Mol Biol 44: 9229.
- Tiringer K, Treis A, Fucik P, Gona M, Gruber S, et al. (2013) A Th17 and Th2skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 187: 6219.
- Tu J, Le GY, Ballard HJ (2010) Involvement of the cystic fibrosis transmembrane conductance regulator in the acidosis-induced efflux of ATP from rat skeletal muscle. J Physiol 588: 4563-4578.
- Vanderstocken G, Van de Paar E, Robaye B, di Pietrantonio L, Bondue B, et al. (2012) Protective role of P2Y2 receptor against lung infection induced by pneumonia virus of mice. PLoS One 7: e50385.
- Kushwah R, Gagnon S, Sweezey NB (2013) Intrinsic predisposition of naïve cystic fibrosis T cells to differentiate towards a Th17 phenotype. Respir Res 14: 138. [Crossref]
- Fukahori S, Matsuse H, Tsuchida T, Kawano T, Nishino T, et al. (2014) Clearance of Aspergillus fumigatus is impaired in the airway in allergic inflammation. Ann Allergy Asthma Immunol 113: 180-186. [Crossref]
- Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, et al. (2011) The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med 184: 252-258. [Crossref]
- Iannitti RG, Carvalho A, Cunha C, De Luca A, Giovannini G, et al. (2013) Th17/Treg imbalance in murine cystic fibrosis is linked to indoleamine 2,3-dioxygenase deficiency but corrected by kynurenines. Am J Respir Crit Care Med 187: 609-620. [Crossref]
- Bacher P, Kniemeyer O, Scho¨nbrunn A, Sawitzki B, Assenmacher M, et al. (2014) Antigenspecific expansion of human regulatory T cells as a major tolerance mechanism against mucosal fungi. Mucosal Immunol 7: 91628.
- Siegmann N, Worbs D, Effinger F, Bormann T, Gebhardt M, et al. (2014) Invariant natural killer T (iNKT) cells prevent autoimmunity, but induce pulmonary inflammation in cystic fibrosis. Cell Physiol Biochem. 34: 5670.
- Singh S, Barr H, Liu YC, Robins A, Heeb S, et al. (2015) Granulocytemacrophage colony stimulatory factor enhances the pro inflammatory response of interferon-α treated macrophages to Pseudomonas aeruginosa infection. PLoS One 10: e0117447.
- Assani K, Tazi MF, Amer AO, Kopp BT (2014) IFN stimulates autophagymediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS One 9: e96681.
- Ni I, Ji C, Vij N (2015) Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PLoS One 10: e0121200. [Crossref]
- McCaslin CA, Petrusca DN, Poirier C, Serban KA, Anderson GG, et al. (2015) Impact of alginate-producing Pseudomonas aeruginosa on alveolar macrophage apoptotic cell clearance. J Cyst Fibros 14: 70-77. [Crossref]
- Lamothe J, MA Valvano (2008) Burkholderia cenocepacia-induced delayof acidification and phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective macrophages. Microbiology 154: 3825-3834.
- Guillemot L, Medina M, Pernet E, Leduc D, Chignard M, et al. (2014) Cytosolic phospholipase A2a enhances mouse mortality induced by Pseudomonas aeruginosa pulmonary infection via interleukin 6. Biochimie 107: 95104.
- Gao Z, Su X (2015) CFTR regulates acute inflammatory responses in macrophages. QJM 108: 951-958. [Crossref]
- Zhang PX, Cheng J, Zou S, D'Souza AD, Koff JL, et al. (2015) Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat Commun 6: 6221. [Crossref]
- Cremer TJ, Shah P, Cormet Boyaka E, Valvano MA, Butchar JP, et al. (2011) Aktmediated proinflammatory response of mononuclear phagocytes infected with Burkholderia cenocepacia occurs by a novel GSK3ß dependent, I?B kinaseindependent mechanism. J Immunol 187: 63543.
- Zughaier SM, Stauffer BB, McCarty NA (2014) Inflammation and ER stress downregulate BDH2 expression and dysregulate intracellular iron in macrophages. J Immunol Res 2014: 140728. [Crossref]
- Grassme´ H (2000) CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa. Science 290: 527-530.
- Yoshida M, Xia Y (2003) Heat shock protein 90 as an endogenous protein enhancer of inducible nitric-oxide synthase. J Biol Chem 278: 36953-36958. [Crossref]
- Zhang Y, Li X, Carpinteiro A, Goettel JA, Soddemann M, et al. (2011) Kinase suppressor of Ras-1 protects against pulmonary Pseudomonas aeruginosa infections. Nat Med 17: 341-346. [Crossref]
- Abdalla MY, Ahmad IM, Switzer B, Britigan BE (2015) Induction of heme oxygenase-1 contributes to survival of Mycobacterium abscessus in human macrophages-like THP-1 cells. Redox Biol 4: 328-339. [Crossref]
- Biswal S, Thimmulappa RK, Harvey CJ (2012) Experimental therapeutics of Nrf2 as a target for prevention of bacterial exacerbations in COPD. Proc Am Thorac Soc 9: 47-51. [Crossref]
- Grumelli S, Corry DB, Song LZ, Song L, Green L, et al. (2004) An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 1: e8.
- Trojanek JB, CobosCorrea A, Diemer S, Kormann M, Schubert SC, et al. (2014) Airway mucus obstruction triggers macrophage activation and matrix metalloproteinase 12dependent emphysema. Am J Respir Cell Mol Biol 51: 70920.
- Woods PS, Tazi MF, Chesarino NM, Amer AO, Davis IC (2015) TGF-β-induced IL-6 prevents development of acute lung injury in influenza A virus-infected F508del CFTR-heterozygous mice. Am J Physiol Lung Cell Mol Physiol 308: L1136-1144. [Crossref]
- Pereira LC, Moreira EA, Bennemann GD, Moreno YM, Buss Zda S, et al. (2014) Influence of inflammatory response, infection, and pulmonary function in cystic fibrosis. Life Sci 109: 306.
- Bratcher PE, Rowe SM, Reeves G, Roberts T, Szul T, et al. (2016) Alterations in blood leukocytes of G551D-bearing cystic fibrosis patients undergoing treatment with ivacaftor. J Cyst Fibros 15: 67-73. [Crossref]
- Fritzsching B, ZhouSuckow Z, Trojanek JB, Schubert SC, Schatterny J, et al. (2015) Hypoxic Epithelial Necrosis Triggers Neutrophilic Inflammation via IL1Receptor Signaling in Cystic Fibrosis Lung Disease. Am J Respir Crit Care Med 191: 90213.
- Pazos MA, Pirzai W, Yonker LM, Morisseau C, Gronert K, et al. (2015) Distinct cellular sources of hepoxilin A3 and leukotriene B4 are used to coordinate bacterialinduced neutrophil transepithelial migration. Immunol 194: 130415.
- Dietert K, Reppe K, Mundhenk L, Witzenrath M, Gruber AD (2014) mCLCA3 modulates IL-17 and CXCL-1 induction and leukocyte recruitment in murine Staphylococcus aureus pneumonia. PLoS One 9: e102606. [Crossref]
- McElvaney OJ, O'Reilly N, White M, Lacey N, Pohl K, Gerlza T, et al. (2015) The effect of the decoy molecule PA401 on CXCL8 levels in bronchoalveolar lavage fluid of patients with cystic fibrosis. Mol Immunol 63: 5508.
- Schilter HC, Collison A, Russo RC, Foot JS, Yow TT, et al. (2015) Effects of an antiinflammatory VAP1/SSAO inhibitor, PXS4728A, on pulmonary neutrophil migration. Respir Res 16: 42.
- Kang JH, Hwang SM, Chung IY(2015) S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signalregulated kinase and nuclear factor? B pathways. Immunology 144: 7990.
- McKeon DJ, Condliffe AM, Cowburn AS, Cadwallader KC, Farahi N, et al. (2008) Prolonged survival of neutrophils from patients with Delta F508 CFTR mutations. Thorax 63: 660-661. [Crossref]
- Ng HP, Zhou Y, Song K, Hodges CA, Drumm ML, et al. (2014) Neutrophil-mediated phagocytic host defense defect in myeloid Cftr-inactivated mice. PLoS One 9: e106813. [Crossref]
- Petreccia DC, Nauseef WM, Clark RA (1987) Respiratory burst of normal human eosinophils. J Leukoc Biol 41: 283-288. [Crossref]
- Kolpen M, Bjarnsholt T, Moser C, Hansen CR, Rickelt LF, et al. (2014) Nitric oxide production by polymorphonuclear leucocytes in infected cystic fibrosis sputum consumes oxygen. Clin Exp Immunol 177: 3109.
- Kragh KN, Alhede M, Jensen PØ, Moser C, Scheike T, et al. (2014) Polymorphonuclear leukocytes restrict growth of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Infect Immun 82: 447786.
- Kettle AJ, Turner R, Gangell CL, Harwood DT, Khalilova IS, et al. (2014) Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur Respir J 44: 1229.
- Pohl K, Hayes E, Keenan J, Henry M, Meleady P, et al. (2014) A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 124: 9991009.
- VegaCarrascal I, Bergin DA, McElvaney OJ, McCarthy C, Banville N, et al. (2014) Galectin9 signaling through TIM3 is involved in neutrophilmediated Gramnegative bacterial killing: an effect abrogated within the cystic fibrosis lung. J Immunol 192: 241831.
- Genestet C, Le Gouellec A, Chaker H, Polack B, Guery B, et al. (2014) Scavenging of reactive oxygen species by tryptophan metabolites helps Pseudomonas aeruginosa escape neutrophil killing. Free Radic Biol Med 73: 40010.
- Andrade A, Valvano MA (2014) A Burkholderia cenocepacia gene encoding a nonfunctional tyrosine phosphatase is required for the delayed maturation of the bacteria containing vacuoles in macrophages. Microbiology 160: 133245.
- De Soyza A, CD Ellis, CM Khan, PA Corris, R Demarco de Hor- maeche (2004) Burkholderia cenocepacia lipopolysaccharide, lipid A, and proinflammatory activity. Am J Respir Crit Care Med
- Saini LS, Galsworthy SB, John MA, Valvano MA (1999) Intracellular survival of Burkholderia cepacia complex isolates in the presence of macrophage cell activation. Microbiology 145: 3465-3475. [Crossref]
- Palfreyman RW, Watson ML, Eden C, Smith AW (1997) Induction of biologically active interleukin-8 from lung epithelial cells by Burkholderia (Pseudomonas) cepacia products. Infect Immun 65: 617-622. [Crossref]
- Schwab U, Abdullah LH, Perlmutt OS, Albert D, Davis CW, et al. (2014) Localization of Burkholderia cepacia complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic mucus. Infect Immun 82: 472945.
- Musson JA, Reynolds CJ, Rinchai D, Nithichanon A, Khaenam P, et al. (2014) CD4+ T cell epitopes of FliC conserved between strains of Burkholderia: macrophages and cystic fibrosis implications for vaccines against melioidosis and cepacia complex in cystic fibrosis. J Immunol 193: 60419.
- Bakala N'Goma JC, Le Moigne V, Soismier N, Laencina L, Le Chevalier F, et al. (2015) Mycobacterium abscessus phospholipase C expression is induced during coculture within amoebae and enhances M. abscessus virulence in mice. Infect Immun 83: 78091.
- Bernut A, Herrmann JL, Kissa K, Dubremetz JF, Gaillard JL, et al. (2014) Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proc Natl Acad Sci U S A 111: E94352.
- Rogers GB, Zain NM, Bruce KD, Burr LD, Chen AC, et al. (2014) A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc 11: 496503.
- Bernard H, Desseyn JL, Bartke N, Kleinjans L, Stahl B, et al. (2015) Dietary pectinderived acidic oligosaccharides improve the pulmonary bacterial clearance of Pseudomonas aeruginosa lung infection in mice by modulating intestinal microbiota and immunity. J Infect Dis 211: 15665.
- Kumar L, Chhibber S, Harjai K (2014) Structural alterations in Pseudomonas aeruginosa by zingerone contribute to enhanced susceptibility to antibiotics, serum and phagocytes. Life Sci 117: 24-32. [Crossref]
- Ratjen F, Döring G, Nikolaizik WH (2001) Effect of inhaled tobramycin on early Pseudomonas aeruginosa colonisation in patients with cystic fibrosis. Lancet 358: 983-4.
- Oliver A, Cantón R, Campo P, Baquero F, Blázquez J (2000) High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288: 1251-1253.
- Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, et al. (2000) Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407: 762-764.
- Valerius NH, Koch C, Høiby N (1991) Prevention of chronic Pseudomonas aeruginosa colonisation in cystic fibrosis by early treatment. Lancet 338: 725-726. [Crossref]
- Gibson RL, Emerson J, Mayer-Hamblett N, Burns JL, McNamara S, et al. (2007) Duration of treatment effect after tobramycin solution for inhalation in young children with cystic fibrosis. Pediatr Pulmonol 42: 610-23.
- Döring G, Conway SP, Heijerman HGM, Hodson ME, Høiby N, et al. (2000) Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. European Respiratory Journal 16: 749-767.
- Fernández-Olmos A, García-Castillo M, Maiz L, Lamas A, Baquero F, et al. (2012) In vitro prevention of Pseudomonas aeruginosa early biofilm formation with antibiotics used in cystic fibrosis patients. International Journal of Antimicrobial Agents 40: 173-176.
- Høiby N (2011) Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med 9: 32. [Crossref]
- Noah TL, Ivins SS, Abode KA, Stewart PW, Michelson PH, et al. (2010) Inhaled versus systemic antibiotics and airway inflammation in children with cystic fibrosis and Pseudomonas. Pediatr Pulmonol 45: 281-290. [Crossref]
- Frederiksen B, Koch C, Høiby N (1997) Antibiotic treatment of initial colonization with Pseudomonas aeruginosa postpones chronic infection and prevents deterioration of pulmonary function in cystic fibrosis. Pediatric Pulmonology 23: 330-335.
- Varshosaz J, Ghaffari S, Mirshojaei SF, Jafarian A, Atyabi F, et al. (2013) Biodistribution of amikacin solid lipid nanoparticles after pulmonary delivery. BioMed Research International.
- Islan GA, Bosio VE, Castro GR (2013) Alginate lyase and ciprofloxacin co-immobilization on biopolymeric microspheres for cystic fibrosis treatment. Macromolecular bioscience 13: 1238-1248.
- Islan GA, Martinez YN, Illanes A, Castro GR (2014) Development of novel alginate lyase cross-linked aggregates for the oral treatment of cystic fibrosis. RSC Advances 4: 11758-11765.
- Bjarnsholt T, Jensen PØ, Rasmussen TB, Christophersen L, Calum H, et al. (2005) Garlic blocks quorum sensing and promotes rapid clearing of pulmonary Pseudomonas aeruginosa infections. Microbiology 151: 3873-3880.
- Beck JM, Young VB, Huffnagle GB (2012) The microbiome of the lung. Transl Res 160: 258-266. [Crossref]
- Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, et al. (2012) The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One 7: e45001. [Crossref]
- Bengmark S (2013) Gut microbiota, immune development and function. Pharmacol Res 69: 87-113. [Crossref]
- Chalermwatanachai T, Velásquez LC, Bachert C (2015) The microbiome of the upper airways: focus on chronic rhinosinusitis. World Allergy Organ J 8: 3. [Crossref]
- Smith PM, Howitt MR, Panikov N (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569-73.
- Ramsey BW, Farrell PM, Pencharz P (1992) Nutritional assessment and management in cystic fibrosis: a consensus report. The Consensus Committee. The American Journal of Clinical Nutrition 55: 108-116.
- Beker LT, Russek-Cohen E, Fink RJ (2001) Stature as a prognostic factor in cystic fibrosis survival. J Am Diet Assoc 101: 438-442. [Crossref]
- Borowitz D, Baker RD, Stallings V (2002) Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 35: 246-259. [Crossref]
- Whitsett JA, Dey CR, Stripp BR, Wikenheiser KA, Clark JC, et al. (1992) Human cystic fibrosis transmembrane conductance regulator directed to respiratory epithelial cells of transgenic mice. Nature Genetics 2: 13-20.
- Rosenfeld MA, Yoshimura K, Trapnell BC, Yoneyama K, Rosenthal ER, et al. (1992) In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium. Cell 68: 143-155. [Crossref]
- Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, et al. (1993) Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 75: 207-216.
- Klink D, Schindelhauer D, Laner A, Tucker T, Bebok Z, et al. (2004) Gene delivery systems-gene therapy vectors for cystic fibrosis. Journal of Cystic Fibrosis 3: 203-212.
- Sinn PL, Anthony RM, McCray PB Jr (2011) Genetic therapies for cystic fibrosis lung disease. Hum Mol Genet 20: R79-86. [Crossref]
- Nayerossadat N, Maedeh T, Ali PA (2012) Viral and nonviral delivery systems for gene delivery. Adv Biomed Res 1: 27. [Crossref]
- John G, Chillappagari S, Rubin BK, Gruenert DC, Henke MO (2011) Reduced surface tolllike receptor4 expression and absent interferon? inducible protein10 induction in cystic fibrosis airway cells. Exp Lung Res 37: 31926.
- Kumar L, Chhibber S, Harjai K (2014) Structural alterations in Pseudomonas aeruginosa by zingerone contribute to enhanced susceptibility to antibiotics, serum and phagocytes. Life Sci 117: 24-32. [Crossref]
- Thursfield RM, Davies JC (2012) Cystic Fibrosis: therapies targeting specific gene defects. Paediatric Respiratory Reviews 13: 215-219.