Abstract
Although epithelial ovarian cancers (EOCs) are initially treated with platinum-based chemotherapy, EOCs vary in platinum responsiveness. Cataloging antineoplastic agents according to their effectiveness against platinum-resistant and platinum-sensitive EOC cell lines is valuable for development of therapeutic strategies to avoid platinum inefficacy and to exploit platinum sensitivity. TOV-21G devoid of FANCF expression, OV-90 and SKOV-3 were employed as examples of platinum-sensitive, platinum-intermediate and platinum-resistant cell lines, respectively. Antineoplastic agents examined included mitomycin C, doxorubicin, etoposide, gemcitabine, chlorambucil, paclitaxel, triapine and X-rays. Their effectiveness against cell lines was analyzed by clonogenic assays. Cytotoxic profiles of mitomycin C and carboplatin were similar, with mitomycin C exhibiting greater potency and selectivity against TOV-21G than carboplatin. Cytotoxic profiles of doxorubicin, etoposide and X-rays overlapped with that of carboplatin, while OV-90 overexpressing Rad51 was more resistant to chlorambucil than SKOV-3. The efficacyof paclitaxel and triapinewas independent of platinum sensitivity or resistance. Consistent with these cytotoxicprofiles, cisplatin/mitomycin C, triapine, and paclitaxel differed in the capacity to inducephosphorylation of H2AX, and produced unique inhibitory patterns of DNA/RNA syntheses in HL-60 human leukemia cells. Paclitaxel and triapine in combination produced additive antitumor effects in M109 murine lung carcinoma. In conclusion, mitomycin C is potentially more effective against Fanconi anemia pathway-deficient EOCs than carboplatin. Doxorubicin and etoposide, because of their overlapping cytotoxic properties with carboplatin, are unlikely to be efficacious against platinum-refractory EOCs. Paclitaxel and triapine are effective regardless of platinum sensitivity status, and promising in combination for both platinum-sensitive and platinum-refractory EOCs.
Keywords
carboplatin, Fanconi anemia pathway, mitomycin C, paclitaxel, platinum responsiveness, triapine
Introduction
Although epithelial ovarian cancer (EOC) occurs with a low incidence, it is labeled“thesilent killer” due to the lack of symptoms until the disease has progressed to an advanced stage. The standard initial management of EOC consists of maximum debulking surgery followed by platinum-based chemotherapy. The majority of patients have a complete response to the first-line therapy. However,patients with advanced disease invariably suffer either platinum-sensitive or platinum-refractoryrecurrence, defined by recurrenceaftermore than, or within, 6 monthsfollowing cessation of the induction therapy, respectively[1]. These clinical manifestations are consistent withheterogeneity of primary EOCs in their response toplatinum.
Cisplatin has been virtually replaced by the equally efficacious, considerably less nephrotoxic analog carboplatin [2]. These agents exert cytotoxicity via platination of DNA to produce intrastrand (>90% of total adducts) and interstrand (a small percentage of total adducts) cross-links [3]. While acquired resistance to cisplatin may involve multifactorial processes[4], several observations indicate that DNA repair capacity in individual tumors plays a critical role in determiningthe inherent sensitivity to platinum compounds. For example,fibroblasts derived from patients with Fanconi anemia (FA) and xerodermapigmentosum (XP) are equally hypersensitive to cisplatin[5].Multiple DNA repair factors define cellular responsiveness to cisplatin[6,7].
HereditaryEOCs with germline BRCA1/2 mutationshave been characterized by platinum sensitivity and improved overall survival [8]. BRCA1 (FANCS) and BRCA2 (FANCD1) belong to a group of at least 16 genes, in which biallelic mutations predispose individuals toFA [9].Moreover, deleterious germline and/or somatic mutations in FA genes such as BRIP1 (FANCJ), PALB2 (FANCN) and RAD51C (FANCO)have been identified inserous as well as non-serous EOCs[10]. The FA pathway is essential for the repair of DNA interstrand cross-links (ICLs). The FA gene products form amulti-subunitmonoubiquitin ligase and coordinateDNA repair processesincluding nucleolytic incision byXPF-ERCC1 and Mus81-Eme1, translesion synthesis and homologous recombination[11].According to the report by The Cancer Genome Atlas (TCGA) Research Network, homologous recombination is defective in about half of high-grade serous EOCs [12].
XP characterized by extreme sensitivity to UV irradiation results from a deficiency in nucleotide excision repair (NER) [13].Eukaryotic NER is divided into global genome NER (GG-NER) and transcription-coupled NER (TC-NER)[14]. Hypersensitivity to cisplatin is confined to TC-NER-defective cells [15]. Moreover, 8% of high-grade serous EOCs from the TCGA dataset have been reported to exhibit NER alterations [16].
In hereditary EOCs caused by heterozygous germline mutations of BRCA1or BRIP, the wild-type allele is lost and the mutated allele is retained in tumor tissue, while normal tissue remains heterozygous for the mutation[17,18]. Thus, those tumors are considered to be DNA repair-deficient, while normal tissue in individuals is DNA repair-proficient. In cell line models for a familial BRCA2 mutation, DLD1-BRCA2Dex11/Dex11 (BRCA2 null) is profoundly hypersensitive to carboplatin, while DLD1-BRCA2Dex11/WT (BRCA2 haplodeficient) is as resistant as DLD1 (BRCA2 diploid)[19]. The differential DNA repair capacity in tumor and normal tissueprovidesa wide therapeuticwindow for ICL-causing agents to exerttumor selectivity.
Patients with EOCs have experienced little improvement in post-diagnosis 1-, 3-, and 5-year survival rates for the past 20 years [20]. Although germline BRCA1/2 mutations are associated with better short-term survival, this advantagefades away over time, ultimately being reversed in BRCA1 carriers[21].Since standard treatment has not advanced beyond platinum-based combination chemotherapy, improvement in therapeuticstrategiesfor both platinum-sensitive and platinum-refractory EOCs is needed. To this end,we have cataloged clinically active antineoplastic agents according to their effectiveness against platinum-sensitive, platinum-intermediate and platinum-resistant EOC cell lines. In this report, we have identifiedmitomycin C as an agentmore potent and selective than carboplatin against an EOC cell line deficient in Fanconi anemia pathway, and paclitaxel and triapineas agents effective regardless of platinum sensitivity status.We believe that these pieces of informationare valuable to exploit platinum sensitivity as well as to avoid platinum inefficacy.
Materials and methods
EOC cell lines and reagents
SKOV-3, OV-90[22] and TOV-21G[22]were purchased from the American Type Culture Collection (ATCC, Manassas, VA) in Jan. 2012, May 2014, and Sept. 2014, respectively. Mutations and epigenetic abnormalities present in these cell lines have been reported[23-26]. HL-60 human myeloid leukemia cells obtained from Dr. Robert C. Gallo in 1980 were authenticated by ATCC in Feb. 2014. All cell lineswere maintained in DMEM/F12 medium supplemented with 10% FBS, 50 units/ml of penicillin, and 50 µg/ml of streptomycin.
Carboplatin (Tocris Bioscience, Bristol, UK),stable in concentrated aqueous solution due to self-association [27], wasdissolved in water at 25 mM and stored at room temperature in the dark. Cisplatin (Sigma-Aldrich, St. Louis, MO),which undergoes relatively slow solvolysis in DMSO [28], was dissolved in 0.9% NaCl at 4 mM, immediately aliquoted and stored at -70°C. Thawed aliquots wereused only once. Stock solutions of doxorubicin hydrochloride (Sigma-Aldrich) and gemcitabine hydrochloride (Sigma-Aldrich) were made in water at 10 mM and stored at -20°C.Triapine (FW: 195), largely insoluble in aqueous media[29], was formulated asanisethionate salt(triapine ITS, FW: 321) in this laboratory to eliminate the use of an organic solvent in animal studies. Stock solutions of mitomycin C (a gift from Kyowa Hakko, Japan), etoposide (Sigma-Aldrich), chlorambucil (Sigma-Aldrich), 1,2-bis(methylsulfonyl)-(2-chloroethyl)hydrazine (90CE)[30] and paclitaxel (Alfa Aesar, Ward Hill, MA) weremade in anhydrous DMSO at 2.5 to 50 mM and stored at -20°C. The stock solutions were further diluted with DMSO and added to cell cultureswith the final DMSO concentrations being less than 0.05%. X-rays were delivered using X-RAD 320 (Precision X-Ray, North Branford, CT).
Clonogenic survival assays and growth inhibition assays
The predetermined number ofcells, which had yielded20 to 150 colonies/well in 6-well plates with various treatments, were allowed to adhere overnight, exposed to cytotoxic agents,and incubated for 13 days for colony propagation.For the guanine O6-chloroethylating agent 90CE [30], O6-benzylguanine, an inhibitor of O6-methylguanine-DNA methyltransferase (MGMT) that repairs the DNA alkyl lesions produced by 90CE, was employed to generate MGMT-abrogated conditions [30].Colonies were stained and processed as previously described[31].Colony forming efficiencies of untreated SKOV-3, OV-90 and TOV-21G cells were approximately 0.3, 0.15 and 0.25, respectively. Growth inhibition assays based on cell count,using HL-60 cells treated with various agents for 3 days, were performed aspreviously described [31].
Preparation of whole cell lysates and western analysis
EOC cells seeded at 1.5 x 106 cells/75 cm2-flask wereallowed to adhere overnight and treated with carboplatin or triapinefor 24 h.Trypsinized cellswere washed oncewith phosphate buffered saline (PBS) and solubilized in 200 µl of Laemmli’s sample buffer at 100oC for 5 min. The lysateswere further passed through a QIAshredder (QIAGEN, Valencia, CA) to shear DNA. The extracts were resolved by 6% or 15% polyacrylamide gel electrophoresis (PAGE).Chemiluminescent images were captured using G:BoxiChemi XR (Syngene, Frederick, MD). Antibodies to the following proteins were purchased:p53 (sc-6243, Santa Cruz Biotechnology, Santa Cruz, CA);BRCA2 (MAB2476, R&D Systems, Minneapolis, MN); Rad51 (MS-988, clone 3C10, Thermo Scientific, Fremont, CA); RPA70 (MS-692, Thermo Scientific), PCNA (MS-106, clone PC10, Thermo Scientific);gH2AX (sc-101696, Santa Cruz);deoxycytidine kinase(MABC188, clone 9D4, EMD Millipore, Temecula, CA);O6-methylguanine-DNA methyltransferase(MAB16200, clone MT 3.1, EMD Millipore).
Measurements of DNA/RNA syntheses andgH2AX formation in HL-60 human leukemia cells
DNA and RNA syntheses were measured by incorporation of [methyl-3H]thymidine and [5-3H]uridine into acid-insoluble fractions as previously described [31]. ForgH2AX analysis, histones were extracted from 3 x 106 intact cells with 0.2 M H2SO4and subjected to western analysis[31].
Treatment of Madison 109 murine lung carcinomain vivo
Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee. M109 cells were implanted by subcutaneous inoculation of 1 x 106 cells in 0.1 ml of PBS into the flank of female CD2F1 mice (Charles River Laboratories, Wilmington, MA). Treatment was initiated at Day 1 post implantation. Tumor volume was estimated from caliper measurements of the length and width using the formula: V = (l x w2)/2.
Statistical and mathematical analyses
Clonogenic and growth inhibition assayswere repeated at least three times. Standard deviations for LC90 and IC50 were typically less than 20% of the mean.LC90 values wereinterpolated from 2 data points intersecting 0.1 cell survivals, assuming the portion between the two points was linear. IC50values were derived from logistic 3-parameter regression analyses using KaleidaGraph software (Synergy Software, Reading, PA).
Results
Diversity in platinum sensitivityamong EOC cell lines
Ihnenet al.[26]have reported that a panel of 39 EOC cell lines is diverse in carboplatin sensitivity in their study offactors affecting the sensitivity to the PARP inhibitor rucaparib. The IC50 (concentration giving 50% inhibition) differential between the most sensitive cell line TOV-21G (0.26 µM) and the nine most resistant cell lines (> 10 µM) is over 38-foldwith the restof the cell linesexhibiting graded sensitivity to carboplatin (Figure 1A). Taniguchi et al.[25] haveuncoveredevidence that the hypersensitivity of TOV-21G cells to cisplatinis due toa disruption of the Fanconi anemia (FA) pathwayresulting froman epigenetic silencing of the FANCF gene via CpG island methylation. We choseTOV-21G (IC50, 0.26 µM), OV-90 (IC50, 1.3 µM) and SKOV-3 (IC50, >10 µM) as examples of platinum-sensitive, platinum-intermediate and platinum-resistant ovarian carcinoma cell lines, respectively. Cell/colony morphology and known characteristics of these cell lines are summarized in Figure 1B.
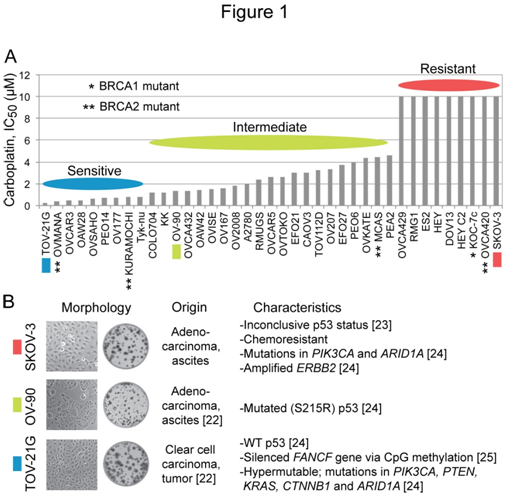
Figure 1. Diversity in carboplatin sensitivity among EOC cell lines, and characteristics of SKOV-3, OV-90 and TOV-21G cells employed in the study. Panel A, the bar graph was generated using the reported IC50 values for carboplatin in a panel of 39 EOC cell lines [26]. Panel B, the information about the origin, phenotypes and mutations in each cell line were collected from ATCC catalog and earlier reports [22-25].
Effectiveness of various antineoplastic agents againstSKOV-3, OV-90 and TOV-21G cells
The three EOC cell lines differing in platinum sensitivity were employedto examine cross-resistance between carboplatin and other clinically active anticancer agents. Figure 2A shows concentration-dependent survival curves for each agent.Values of LC90(concentration giving 90% lethality)together withvalues of fold resistance (degree of resistance in OV-90 and SKOV-3 relative to TOV-21G) in parentheses are summarized in Figure 2B.
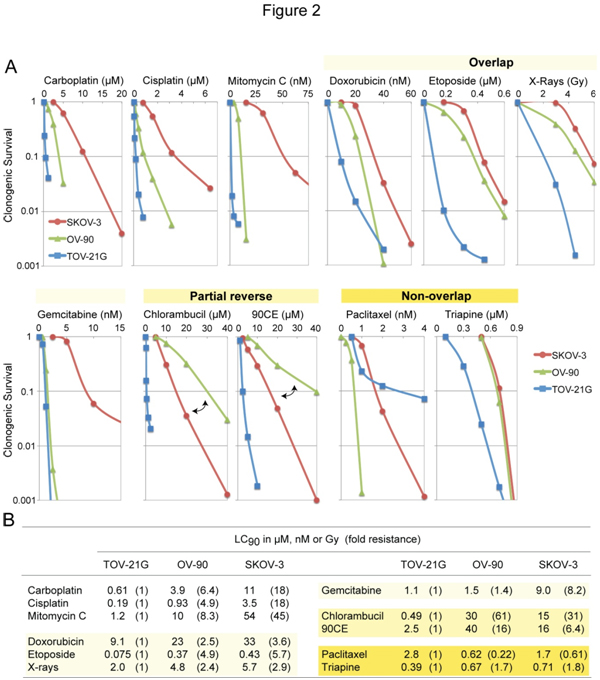
Figure 2. Cytotoxic profiles of various antineoplastic agents against SKOV-3, OV-90 and TOV-21G cells. Panel A, cytotoxicity was measured using clonogenic assays. For the guanine O6-chloroethylating agent 90CE, cells were pretreated with 10 µM O6-benzylguanine for 2 h to abrogate activity of O6-methylguanine-DNA methyltransferase (MGMT). O6-Benzylguanine was present in the medium throughout treatment and colony propagation. Panel B, for clarification, values of LC90 (concentration giving 90% lethality) and fold resistance (degrees of resistance in OV-90 and SKOV-3 relative to TOV-21G) are tabulated.
Although carboplatinreacts with DNA much more slowlythancisplatin, the two analogs have been shown to generate nearly identical arrays of DNA adducts[32] and to produce similar resistance profiles[33]. Consistent with those findings, the present study confirms that carboplatin and cisplatin produce highly comparable resistance profiles(Figure 2A). Amongantineoplastic agents examined, the sensitivity profile produced by mitomycin C was most akin to that produced by carboplatin (Figure 2A). Moreover, mitomycin C exhibited greater degrees of potency (reflected in LC90 in nM versus µM for carboplatin, Figure 2B) as well as selectivity(reflected in largervalues of fold resistance, Figure 2B).
Relative to carboplatin/mitomycin C, antineoplastic agentsproduced sensitivity profiles collectively in overlapping, partially reversed, and non-overlappingmanners(Figure 2A). Thus, doxorubicin, etoposide, X-rays and gemcitabineproduced overlapping profiles with ashared rank order of TOV-21G <OV-90 <SKOV-3.The nitrogen mustard chlorambucil and the guanine O6-chloroethylating agent 90CE [30] produced partially reversed profiles in that the platinum-intermediate OV-90 was more resistant to these agents than the platinum-resistant SKOV-3.Paclitaxel produced a unique rank order of OV-90 <SKOV-3 <TOV-21G. Triapine produced a rank order of TOV-21G <OV-90 = SKOV-3 withthe degree of resistance in SKOV-3 relative to TOV-21G being less than two-fold (Figure 2B). Therefore, relative to carboplatin/mitomycin C, both paclitaxel and triapineproduced sensitivity profiles essentially in a non-overlapping manner.
Untreated and drug-treated levels ofseveral DNA repair/replicationproteins
SKOV-3, OV-90 and TOV-21G cells were untreated or treated with 10 µM carboplatin or 1 µM triapine for 24 h. Then, whole cell extracts were prepared and subjected to western analysis(Figure 3). Stabilization of BRCA2 andRPA70 (less noticeable)occurredfollowingtriapinetreatment.The notabledifferenceamongthe three cell lines wasoverexpression ofRad51recombinasein OV-90 cells (Figure 3). After triapine treatment, phosphorylation of H2AX (gH2AX)occurredprominently in TOV-21G. The PCNA levelwasalmost constantregardless of cell line and treatment, and served as a loading control.
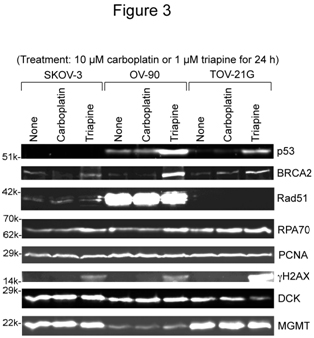
Figure 3. Western analyses of relevant DNA damage response proteins following treatment with carboplatin or triapine. Whole cell extracts were prepared from SKOV-3, OV-90 and TOV-21G cells untreated, or treated with 10 µM carboplatin or 1 µM triapine for 24 h. BRCA2, p53, and RPA70 were resolved by 6% PAGE. Rad51, PCNA, gH2AX, DCK and MGMT were resolved by 15% PAGE. RPA70, replication protein A 70 kDa DNA-binding subunit; PCNA, proliferating cell nuclear antigen; DCK, deoxycytidine kinase; MGMT, O6-methylguanine-DNA methyltransferase.
2021 Copyright OAT. All rights reserv
In regard to p53 status, TOV-21Gexhibitedthe wild-type characteristics, i.e., an undetectable level in untreated cells and accumulation in triapine-treated cells. The reported S215R mutation in OV-90 cells[24]together with prominent accumulationfollowingtriapine treatment (Figure 3) suggests that OV-90 cells contain both wild-type and mutatedTP53genes. The absence of p53 in SKOV-3 cells underboth untreatedandtriapine-treated conditions suggeststhat this cell line lacks functionalTP53 genes.
SKOV-3 was6- to 8-fold more resistant to gemcitabine than OV-90 and TOV-21G cells (Figure 2B).The greater level of deoxycytidine kinase (DCK, responsible for the initial phosphorylation of gemcitabine) in SKOV-3 (Figure 3) precludes alack of the activation step as a mechanism of resistance.
Inhibition of DNA/RNA syntheses and phosphorylation of H2AX by carboplatin/mitomycin C, triapine and paclitaxel
Cytotoxic activity of the DNA interstrand cross-linking agentscisplatin and mitomycin Cis attributed to interference with DNA replication and/or transcription[4], while cytotoxic activities of triapine and paclitaxel were independent of platinum sensitivity/resistance (Figure 2A and 2B). To gain mechanistic insight into theseunique cytotoxic properties, thecapacity of these agents to inhibit DNA and RNA syntheses after short-term (2 h) and long-term (18 h) treatment was measured using HL-60 human leukemia cells. For each agent, the lowest concentrationchosen for treatment was near its respective IC50 value, predetermined by growth inhibition assays (Figure 4A).
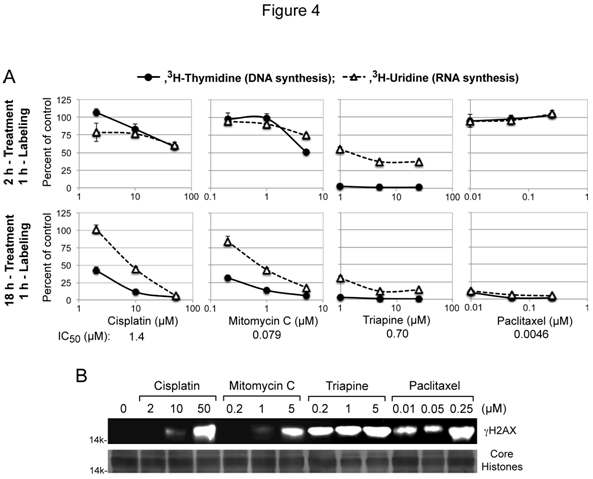
Figure 4. Inhibition of DNA/RNA syntheses and induction of gH2AX by cisplatin, mitomycin C, triapine or paclitaxel. Panels A, for short-term treatment, HL-60 cells (2 x 106 cells/ml) were treated with various agents for 2 h, and labeled with 3H-thymidine or 3H-uridine for 1 h. For long-term treatment, HL-60 cells (4 x 105 cells/ml) were treated with various agents for 18 h, condensed to 2 x 106 cells/ml and labeled with 3H-thymidine or 3H-uridine for 1 h. Panels B, HL-60 cells (4 x 105 cells/ml) were treated with various agents for 18 h. Histones extracted from intact cells were subjected to 15% PAGE and western analyses using an anti-gH2AX antibody. For a loading control, duplicate gels were used to stain core histones with GelCode Blue Safe Protein Stain (Thermo Scientific).
Cisplatin and mitomycin C produced nearly identicalinhibitorypatterns, preferentially inhibiting DNA synthesis only after 18 h treatment (Figure 4A). In contrast, triapine caused immediate complete cessation of DNA synthesis asshown in the short-term (2 h) treatment (Figure 4A).Effects of paclitaxel on nucleic acid synthesesyet differed from those oftriapine and cisplatin/mitomycin C. Thus, uninterruptednucleic acid synthesesafter2 htreatment with paclitaxel were followed by complete shutdown of both DNA and RNA syntheses after 18 h treatment (Figure 4A).
DNA double-stand breaks induce rapid phosphorylation of histone H2AX(gH2AX)in a chromatin microenvironment[34]. The capacity of cisplatin/mitomycin C, triapine and paclitaxelto inducegH2AX formationwasexamined in HL-60 cells treated with these agents for 18 h (Figure 4B).Triapine caused pronounced gH2AX formation at a concentration (0.2 µM) 3.5 fold lower than its IC50 value. In contrast, significant H2AX phosphorylationwas induced by cisplatin/mitomycin C at concentrations 40 to 60 fold higher than their IC50 values. Paclitaxel,which does not directly targetDNA,also caused significantgH2AXaccumulation.These results collectively indicate that phosphorylation of H2AX is inadequate to be a cytotoxic biomarker.
Antineoplastic agents cataloged by their effectiveness against platinum-sensitive and platinum-resistant cell lines and underlying mechanisms of action
Based on the survival curves, and LC90 values presented in Figure 2A and 2B, anticancer agents werecatalogedaccording to their effectiveness against platinum-sensitive TOV-21G, platinum-intermediate OV-90and platinum-resistant SKOV-3 (Figure 5A).Mitomycin C stands out for its effectiveness against FANCF-deficient TOV-21G with greater potency and selectivity than carboplatin, while paclitaxel and triapine are prominent for their effectiveness against bothplatinum-sensitive and platinum-resistant EOC cells. Figure 5B summarizesthe primary sites of action and cytotoxic mechanismsofcarboplatin/mitomycin C, triapine and paclitaxel that form the basis of theirunique cytotoxic properties.This issue is addressedfurther in the discussion section.
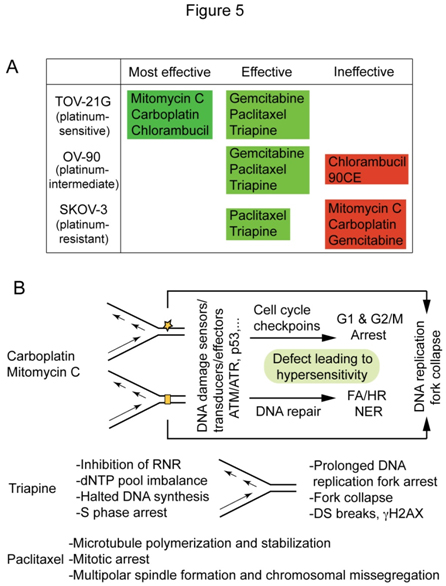
Figure 5. Antineoplastic agents cataloged according to their effectiveness against TOV-21G, OV-90 and SKOV-3 (A) and distinguishable cytotoxic mechanisms of carboplatin/mitomycin C, triapine and paclitaxel (B).
Antitumor activity of paclitaxelalone or incombination with triapinein Madison 109 murine lung carcinoma
Effectiveness of triapine alone or in combination with cisplatin, doxorubicin or etoposide has previously been demonstrated in the EOC cell line A2780 xenograft tumor model [35]. In this study, antitumor activity of triapine alone or in combination with paclitaxel was examined in M109 lung carcinoma, a murine syngeneic tumor model weakly responsive to cisplatin[36]. Figure 6 shows that triapine ITS (water soluble isethionate salt form of triapine) is as effective as paclitaxel in producing tumor growth delay. Furthermore, a combination of paclitaxel with triapine resulted in extensive tumor growth delay in an additive manner.
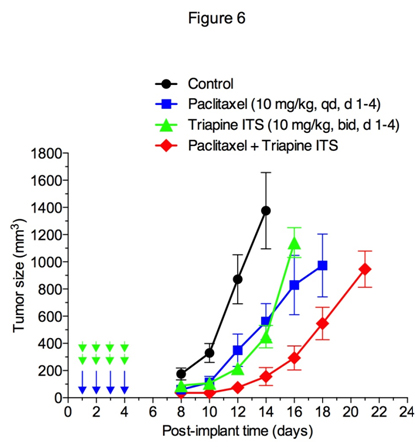
Figure 6. Antitumor activity of paclitaxel and triapine, alone or in combination, against Madison 109 murine lung carcinoma. Tumor-bearing mice (5 mice/group) were treated with i.p. injection of paclitaxel at a dose of 10 mg/kg once a day for 4 days, triapine isethionate salt (triapine ITS) at a dose of 10 mg/kg (equivalent to 6 mg/kg of triapine) twice a day, 8 h apart, for 4 days, or combination of both. Paclitaxel dissolved in DMSO at 10 mg/ml was injected in a volume of 10 µl/10 g weight using insulin syringes. Triapine ITS dissolved in water at 1 mg/ml (pH 4.5) was neutralized with 2 molar equivalents of NaHCO3 immediately before injection.
Discussion
Although platinum-containing regimens have been the mainstay of initial treatment forEOCs,cancer recurrence iseither platinum-refractory or platinum-sensitive. Thus, it is essential(a) to comprehendthe mechanismsunderlyinginherent platinum resistance andsensitivity, (b) to develop means to distinguish between platinum-refractoryand platinum-sensitive EOCs prior to chemotherapy, and(c) to applyappropriate treatment to platinum-refractory and platinum-sensitive EOCs to avoid platinum inefficacy and to exploit platinum sensitivity, respectively.A PubMed search for resistance to cisplatin, sensitivity to cisplatin, and mechanism of action of cisplatinreturns ~8,400, ~5,500, and ~46,000 articles, respectively. Despite thisvast body of research, there remains much to be clarified about the nature ofDNA adducts (DNA intrastrand or interstrand cross-links, or both) crucial for their cytotoxicity,the types of DNA damage repair systems involved in removal of critical DNA adducts, andthechain of events in which inhibition of DNA/RNA syntheses results in ultimate cell death.
The striking diversity of carboplatin sensitivity in a panel of 39 EOC cell lines derived from serous and non-serous EOCs (Figure 1A) reported by Ihnenet al. [26] is in contrast with the modestly variable platinum sensitivity in the NCI-60, a panel of 60 cell lines derived from diverse human cancers including six ovarian carcinomas (NCI DPT Data). Among the 39 EOC cell lines (Figure 1A), 18 cell lines (46%) are markedly sensitive to carboplatin (IC50 values less than 2 µM). The existence of a large fraction ofplatinum-hypersensitive EOC cell lines is compatible with a high mutation rate in DNA repair pathwaysinvolving HR, FA and NER found in human primary EOCs [10,12,16]. Thus, it is of great interest to determine the molecular mechanisms underlyingplatinum-hypersensitivity of theseEOC cell lines,and their susceptibility to a spectrum of antineoplastic agents.
In this study, we used SKOV-3, OV-90 and TOV-21G as examples of platinum-resistant, platinum-intermediate and platinum-sensitive EOC cell lines, respectively. Based on the genomic profiles of primary high-grade serous ovarian carcinoma (HGSOC)andpermanentEOCcell lines, OV-90 and SKOV-3 are possibly and unlikely, respectively, derived from HGSOC[24]. TOV-21G,established from a clear cell carcinoma [22],is known to be deficient in FANCF expression due to gene silencing, and hypersensitive to cisplatin[25].
Cellular hypersensitivity to DNA interstrand cross-linking agents is one of the defining characteristics of FA [11]. FANCF-deficient TOV-21G consistently exhibited profound hypersensitivity to mitomycin C (LC90, 1.2 nM), chlorambucil (LC90, 0.49 µM) and carboplatin (LC90, 0.61 µM). TOV-21G was 45-, 31-, and 18-fold more sensitive to mitomycin C, chlorambucil and carboplatin, respectively, than platinum-resistant SKOV-3. The greater potency (LC90 at nM) and selectivity (fold sensitivity) exhibited by mitomycin C suggest potential utility of this agent for FA pathway-deficient EOCs.
It is noteworthy that the platinum-intermediate OV-90 was more resistant to chlorambucil and 90CE than the platinum-resistant SKOV-3 (partial reverse, Figure 2A). Although cisplatin, mitomycin C, chlorambucil and 90CEall causeDNA interstrand cross-links (ICLs), chemical structures of ICL adducts produced by individual agentsare quite distinct[3,30,37-39]. Yet,repair mechanisms for the ICLs produced bycisplatin/mitomycin C on one hand, and chlorambucil/90CE on the other,appear to be distinguished. Western analyses of several key DNA damage repair proteins (Figure 3) clearlyindicated that OV-90 cells overexpress Rad51. Since Rad51 shares amino acid sequence similarity and domain structures with Rad51 paralogs (Rad51B, Rad51C, Rad51D, XRCC2, XRCC3 and meiosis-specific DMC1)[40], involvement ofRad51 paralogsin the repair of various types of ICLsneeds to be clarified in the future.
It is also noteworthy that gemcitabine was equally cytotoxic to the platinum-sensitive TOV-21G and platinum-intermediate OV-90. Thus, unlike doxorubicin, etoposide and X-rays whose cytotoxic profiles overlapped with that of carboplatin (Figure 2A), the cytotoxic property of gemcitabine appeared to be unique. In support of this notion,Jones et al. [41] have demonstrated thatthe presence of functional BRCA2 and Rad51 promotes (rather than decreases) the cytotoxic activity of gemcitabine.Theseobservationsaltogether suggest aneffectiveness of gemcitabinefor an undefined subpopulation of EOCs.
One of the important findings in this report is the efficacy of paclitaxel and triapine against EOC cell lines regardless of platinum sensitivity status, suggesting usefulness of these agents in the treatment of both platinum-sensitive and platinum-refractory EOCs.An inverse relationship between platinum and taxenewith regard totheirclinical efficacyagainstEOCsandtheir cytotoxic activityagainstEOC cell lines has long been recognized and extensively reviewed[42].In light oftheobvious disparityin the mode of action of these two agents(Figure 5B), it isconceivable that the cytotoxic activity of paclitaxel is independent of platinum resistance or sensitivity. On the other hand, triapine and carboplatin are agents bothknown to cause DNA replication stress.As inhibitors of ribonucleotidereductase, triapine is200 to 300 fold more potent than hydroxyurea[31],an agenthistoricallyemployed to generateDNA replication stress in cultured cells[43]. The sequential events in which imbalance of dNTP pools caused by ribonucleotidereductase inhibitionis ultimately conveyed to DNA double strand breaks are shown in Figure 5B. Carboplatin and mitomycin Cdiffer from triapine in that these agents, via direct modifications of DNA structure, elicit DNA damage response pathways that protect cells from deleterious genotoxicity(Figure 5B).Thus, a defect in DNA damage response pathways results in cellular hypersensitivity to these agents,whereas extensive DNA double-stand breaks by triapineare an ultimate consequence secondary to prolonged DNA replication stress (Figure 5B).
Triapine, currently being evaluated as an antitumor agent in NIH-funded phase I/II clinical trials, was designed and synthesized in the late Dr. Sartorelli’s laboratory [29].Triapine was chosen for clinical development because of its broad-spectrum antitumor activity alone or in combination with cisplatin, doxorubicin, or etoposide in preclinical models [35]. Studies using cultured cells reveal that triapine at concentrations of 0.4 to 0.8 µMproduces immediate cessation of DNA replication due to unbalanced dNTP pools caused by inhibition of ribonucleotidereductase[31].Despite instantaneous effect on DNA synthesis, effective cell-kill by triapine in cultured cells requires prolonged(16 h) drug-exposure, presumably because inhibition of DNA synthesis is reversible upon triapineremoval, and DNA double-strand breaks occur after prolonged DNA replication stress [31]. After a single injection of triapine into L1210 tumor-bearing mice, DNA synthesis recovers faster in normal tissue such as the bone marrow and duodenum,than in tumor cells [35].Consistent with these observations, antitumor activity of triapineis schedule-dependent in preclinical models. Thus,a twice-a-day regimen (6 mg/kg twice a day, 8 h apart, for 5 days for a total dose of 60 mg/kg) is vastly superior to an almost ineffectiveonce-a-day regimen (24 mg/kg once a day for 5 days for a totaldose of 120 mg/kg) that delivers double the total dose [35].
The majority of clinical trials for triapinehave employed asingle 2-houri.v. infusion of triapine per day at doses up to 120 mg/m2, the regimens determined from phase I trials as tolerable doses, whichmaynot be therapeutically optimum[31]. Split-doseregimens thatkeep plasma concentrations of triapine low and sustain DNA replication stress for a prolonged period of time, should be explored in clinical trials. Furthermore, low but sustained concentrations of triapine are unlikely to cause host toxicity such as methemoglobinemia and hypoxia.
In this study, we cataloged antineoplastic agents according to the effectiveness against platinum-sensitive TOV-21G, platinum-intermediate OV-90 and platinum-resistance SKOV-3. Overall, themechanisms surrounding platinum sensitivity/resistance are multifactorial. Thus, straightforward approaches to personalized medicineshould incorporate ex-vivo drug sensitivity assaysin which sensitivity/resistance of individual EOC sample to an array of antineoplastic agents can be tested. Prerequisites for valid assays include a) enrichment of EOC cells via combined positive (EpCAM beads) and negative (anti-fibroblast beads) selections, b) provision of growth-stimulating environments to enhance the sensitivity to cell-replication-targeting antineoplastic agents, c) use of appropriate cellular markers to distinguish tumor from normal cells, and d) use of adequate cytotoxic markers to detect cytotoxicity. Efforts to establish such assays are critical for successful personalized medicine.
Authorship
K.I. designed and developed the concept of this work, conducted the experiments, analyzed the data and drafted the paper. Y-L.Z., Z-P.L. and R.Z. conducted the experiments and analyzed the data. P.G.P., K.S., R.P.B., T.J.R. and A.C.S. contributed to the concept and design of this work, analyzed the data, and polished the report. E.S.R. contributed to the concept and design of this work, provided analysis of the data, polished the report, obtained funding and gave final approval.
Acknowledgement
We are grateful to Dr. Bret Halpern for his helpful discussions.
Funding
This work was supported in part by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant K12HD047018 (E.S.R.) and a grant from theDiscovery to Cure to E.S.R.
References
- Banerjee S, Bookman M, Gore M (2011) Systemic therapy for ovarian cancer, current treatment, recent advances, and unmet needs. Emerging Therapeutic Targets in Ovarian Cancer. Springer, London: 1–33.
- Kelland LR (2005) Emerging drugs for ovarian cancer. Expert Opin Emerg Drugs 10: 413-424. [Crossref]
- Jamieson ER, Lippard SJ (1999) Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 99: 2467-2498. [Crossref]
- Jung Y, Lippard SJ (2007) Direct cellular responses to platinum-induced DNA damage. Chem Rev 107: 1387-1407. [Crossref]
- Plooy AC, Van Dijk M, Berends F, Lohman PH (1985) Formation and repair of DNA interstrand cross-links in relation to cytotoxicity and unscheduled DNA synthesis induced in control and mutant human cells treated with cis-diamminedichloroplatinum(II). Cancer Res 45: 4178-4184. [Crossref]
- Zdraveski ZZ, Mello JA, Marinus MG, Essigmann JM (2000) Multiple pathways of recombination define cellular responses to cisplatin. Chem Biol 7: 39-50. [Crossref]
- Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, et al. (2005) Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res 65: 11704-11711. [Crossref]
- Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, et al. (2012) Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 307:382-390. [Crossref]
- Wang AT, Smogorzewska A (2015) SnapShot: Fanconi anemia and associated proteins. Cell 160: 354-354.e1. [Crossref]
- Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, et al. (2014) Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 20: 764-775. [Crossref]
- Kim H, D'andrea AD (2012) Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26: 1393-1408. [Crossref]
- Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609-615. [Crossref]
- Lehmann AR (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85: 1101-1111. [Crossref]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465-481. [Crossref]
- Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, et al. (2002) Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res 62: 4899-4902. [Crossref]
- Ceccaldi R, O'connor KW, Mouw KW, Li AY, Matulonis UA, et al. (2015) A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res 75: 628-634. [Crossref]
- Neuhausen SL, Marshall CJ (1994) Loss of heterozygosity in familial tumors from three BRCA1-linked kindreds. Cancer Res 54: 6069-6072. [Crossref]
- Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, et al. (2011) Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet 43: 1104-1107. [Crossref]
- Hucl T, Rago C, Gallmeier E, Brody JR, Gorospe M, et al. (2008) A syngeneic variance library for functional annotation of human variation: application to BRCA2. Cancer Res 68: 5023-5030. [Crossref]
- Vaughan S, Coward JI, Bast RC, Jr., Berchuck A, Berek JS, et al. (2011) Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer 11: 719-725.
- Candido-Dos-Reis FJ, Song H, Goode EL, Cunningham JM, Fridley BL, et al. (2015) Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin Cancer Res 21: 652-657. [Crossref]
- Provencher DM, Lounis H, Champoux L, Tetrault M, Manderson EN, et al. (2000) Characterization of four novel epithelial ovarian cancer cell lines. In Vitro Cell Dev Biol Anim 36: 357-361. [Crossref]
- Berglind H, Pawitan Y, Kato S, Ishioka C, Soussi T (2008) Analysis of p53 mutation status in human cancer cell lines. Cancer Biol Ther 7: 701-710. [Crossref]
- Domcke S, Sinha R, Levine DA, Sander C, Schultz N (2013) Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 4: 2126-2135.
- Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, et al. (2003) Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 9: 568-574. [Crossref]
- Ihnen M, Zu Eulenburg C, Kolarova T, Qi JW, Manivong K, et al. (2013) Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther 12: 1002-1015. [Crossref]
- Di Pasqua AJ, Kerwood DJ, Shi Y, Goodisman J, Dabrowiak JC (2011) Stability of carboplatin and oxaliplatin in their infusion solutions is due to self-association. Dalton Trans 40: 4821-4825. [Crossref]
- Sundquist WI, Ahmed KJ, Hollis LS, Lippard SJ (1987) Solvolysis reactions of cis- and trans-diamminedichloroplatinum(II) in dimethyl sulfoxide. Structural characterization and DNA binding of trans-[Pt(NH3)2(Me2SO)Cl]+. Inorg Chem 26: 1524-1528.
- Liu MC, Lin TS, Sartorelli AC (1992) Synthesis and antitumor activity of amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J Med Chem 35: 3672-3677. [Crossref]
- Ishiguro K, Lin ZP, Penketh PG, Shyam K, Zhu R, et al. (2014) Distinct mechanisms of cell-kill by triapine and its terminally dimethylated derivative Dp44mT due to a loss or gain of activity of their copper(II) complexes. Biochem Pharmacol 91: 312-322. [Crossref]
- Ishiguro K, Zhu YL, Shyam K, Penketh PG, Baumann RP, et al. (2010) Quantitative relationship between guanine O6-alkyl lesions produced by onrigin and tumor resistance by O6-alkylguanine-DNA alkyltransferase. Biochem Pharmacol 80: 1317-1325. [Crossref]
- Knox RJ, Friedlos F, Lydall DA, Roberts JJ (1986) Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res 46: 1972-1979. [Crossref]
- Rixe O, Ortuzar W, Alvarez M, Parker R, Reed E, et al. (1996) Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute's Anticancer Drug Screen panel. Biochem Pharmacol 52: 1855-1865. [Crossref]
- Turinetto V, Giachino C (2015) Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res 43: 2489-2498.
- Finch RA, Liu M, Grill SP, Rose WC, Loomis R, et al. (2000) Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem Pharmacol 59: 983-991. [Crossref]
- Rose WC (1981) Evaluation of Madison 109 lung carcinoma as a model for screening antitumor drugs. Cancer Treat Rep 65: 299-312. [Crossref]
- Tomasz M (1995) Mitomycin C: small, fast and deadly (but very selective). Chem Biol 2: 575-579. [Crossref]
- Gruppi F, Hejazi L, Christov PP, Krishnamachari S, Turesky RJ, et al. (2015) Characterization of nitrogen mustard formamidopyrimidine adduct formation of bis(2-chloroethyl)ethylamine with calf thymus DNA and a human mammary cancer cell line. Chem Res Toxicol 28: 1850-1860. [Crossref]
- Deans AJ, West SC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11: 467-480.
- Suwaki N, Klare K, Tarsounas M (2011) RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol 22: 898-905. [Crossref]
- Jones RM, Kotsantis P, Stewart GS, Groth P, Petermann E (2014) BRCA2 and RAD51 promote double-strand break formation and cell death in response to gemcitabine. Mol Cancer Ther 13: 2412-2421. [Crossref]
- Stordal B, Pavlakis N, Davey R (2007) A systematic review of platinum and taxane resistance from bench to clinic: an inverse relationship. Cancer Treat Rev 33: 688-703. [Crossref]
- Fugger K, Chu WK, Haahr P, Kousholt AN, Beck H, et al. (2013) FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat Commun 4: 1423-1430. [Crossref]