Amyloidosis includes a spectrum of diseases that involve the deposition of misfolded proteins in various tissues throughout the body. There are many different proteins known to cause amyloidosis and the etiology of the amyloid subtype determines where the abnormal proteins deposit. AL, ATTRwt (formerly senile systemic amyloidosis), and ATTRm (formerly familial amyloidosis) are the most common amyloid subtypes that infiltrate cardiac tissue, resulting in a restrictive cardiomyopathy. In this series we report one case of each cardiac amyloidosis subtype, AL, ATTRwt, and ATTRm, that presented to Harbor-UCLA Medical Center between 2012-2013. All three patients had biopsies confirming cardiac amyloidosis as well as a cardiac MRI. Unfortunately, two of the three patients with cardiac amyloidosis passed away from PEA arrest illustrating the high mortality rate for this disease process. Since early diagnosis and treatment has shown improved median survival rates, we aim to review the diagnostic evaluation, treatment approaches, and prognosis for AL, ATTRwt, and ATTRm cardiac amyloidosis.
amyloid, cardiac amyloidosis, diastolic heart failure
We report three cases of cardiac amyloidosis that presented to our institution, each with different subtypes and pathology. The aim of this review article is to highlight the clinical presentation of cardiac amyloidosis, as well as the diagnosis and treatment of each subtype. Unfortunately this patient group has a high mortality, but through accurate diagnosis and timely work-up some morbidity may be diminished.
Amyloidosis results from the abnormal folding of proteins into insoluble beta pleated sheets. These amyloid deposits can be identified by their apple-green birefringence under polarized light when stained by Congo red, or positive staining with sulfated Alcian blue. Amyloid deposition can occur from both inherited and non-inherited causes and has variable cardiac penetrance depending on the underlying cause.
Classically, amyloidosis was first described in patients with refractory systemic illness, likely the result of serum amyloid A protein, an acute phase reactant [1]. This type of amyloidosis, amyloid A (AA), predominately affects the kidneys and the heart less than 10% of the time [1,2].
The most common type of amyloidosis results from monoclonal immunoglobulin deposition by clonal B cell lymphoproliferative diseases is known as AL amyloidosis, and has been historically termed primary amyloidosis. Often caused by low-grade plasma cell dyscrasias, the immunoglobulin light chains deposit as fibrils in multiple tissues, including the heart in about half of the cases [1,3].
Familial amyloidosis, formally known as ATTRm, is the result of mutations on exons 1-4 of chromosome 18, resulting in one of 100 polymorphisms of the alpha transthyretin protein (TTR) [1]. ATTRm is autosomal dominant, predominately affecting the heart and nervous system, with varying penetrance depending on the mutation. ATTRm is confirmed on biopsy with a positive immunohistochemical staining pattern for transthyretin and a positive gene screening for the transthyretin gene TTR [1,2].
ATTRwt amyloidosis, formally known as Senile systemic amyloidosis, results in the misfolding of wild-type transthyretin and commonly involves the heart. As its previous name suggests, ATTRwt is age related, prevalent in men older than 70 years old [1,3]. Unlike ATTRm, ATTRwt will have a negative TTR gene evaluation [1].
Lastly, isolated atrial amyloidosis (IA) results in the misfolding of atrial natriuretic peptide and by definition involves the heart [2] (Table 1).
Table 1. Typical features of the different amyloid types.
Amyloid Type |
Other Names |
Presenting Decade |
Pathophysiology |
Organs Involved |
AA |
Secondary/
reactive |
Variable |
Chronic inflammatory conditions, malignancy |
Kidney, rarely involves the heart |
AL |
Primary |
6th or 7th decade |
Clonal plasma cell derangement |
Heart, kidney, liver, soft tissue, peripheral nervous system |
ATTRm |
Familial Amyloidosis |
6th decade or older |
Mutant transthyretin, apolipoprotein mutations |
Multiorgan involvement, including heart, nervous system, liver, and kidneys |
ATTRwt |
Senile Systemic Amyloidosis |
Typically 8th decade or older |
Wild type transthyretin mutation |
Almost always involves heart, can involve carpal tunnel. |
Isolated atrial amyloidosis (IA) |
|
Typically 8th decade or older |
Atrial natriuretic peptide deposition |
By definition involves the heart |
The epidemiology of cardiac amyloidosis is difficult to ascertain with certainty because cardiac involvement of amyloidosis is rare, affecting 6-10 cases per million individuals in the United States. However, this is thought to be the result of signficant under diagnosis and as improved non-invasive diagnostic approaches arise, this number is likely to increase [3]. In one recent analysis, ATTRwt occurs in greater than 25% of individuals at autopsy but the clinical significance of this finding remains unclear [4,5]. In an often cited but likely regionally, ethnic diversity limited survey, the age-adjusted incidence of AL amyloidosis is estimated to be beetween 5.1 and 12.8 per million person years [6].
In this case series, we present three individuals, one case of AL, a case of suspected ATTRwt, and a case of likely ATTRm, who all presented to a Harbor-UCLA Medical Center in Torrance, California.
Cardiac involvement of amyloidosis presents mainly as a restrictive cardiomyopathy, with initial diastolic dysfunction that can progress to biventricular systolic and diastolic dysfunction. Accordingly, presenting symptoms are typical of heart failure and include fatigue, exertional dyspnea, and edema [1,3]. As the heart becomes progressively larger and as the amyloid deposits into the heart’s conduction system, patients are prone to arrythmias, especially atrial fibrillation [1,5]. Sudden cardiac death, often from ventricular and PEA rhythms, have also been associated [3].
A 63 year old male with known hypertension, presented to our hospital with progressive shortness of breath and fatigue for one year, which had worsened over the previous four days. He endorsed exertional chest pain, orthopnea, paroxysmal nocturnal dyspnea, and bilateral lower extremity swelling. Non-cardiac symptoms included a chronic diarrhea and some mild foaming of his urine.
He had been previously worked up inconclusively at an outside hospital. Of note, he was found to have an abnormal serum light chains, with lambda light chains measuring 130mg/dL, kappa light chains measuring 582mg/dL, total protein of 4.6g/dL, and albumin of 2.9g/dL. His 24 hour urine collection was not suggestive of nephrotic range proteinuria. An outside hospital echocardiogram had shown diastolic dysfunction heart failure with preserved left ventricle function (estimate LVEF 65-70%). It was noted to have evidence of a restrictive cardiomyopathy. The patient's PMIBI was without evidence of infarct. Given his abnormal serum light chains, the patient had a skull series x-ray examination which did not demonstrate any lytic lesions.
Upon presentation to our hospital, the patient had signs of volume overload, including jugular vein distention roughly 1/3 the height of the patient’s neck, decreased breath sounds on both bases, and bilateral pitting lower extremity edema. His electrocardiogram (ECG) demonstrated a low voltage pseudo-inferior infarct pattern (Figure 1). He also had consistently elevated troponin-I's, ranging between 0.11 and 0.22ng/dL. A chest x ray and CT chest with contrast revealed large bilateral pleural effusions, without evidence of infiltrative disease or pulmonary embolus. The patient subsequently had several therapeutic thoracenteses, which showed transudative effusions (pleural fluid lactate dehydrogenase 86U/L, serum lactate dehydrogenase 155U/L, pleural protein 2.7g/dL, serum protein 4.8g/dL). Given the high suspicion for cardiac amyloidosis, the patient had a cardiac MRI with and without gadolinium, which showed normal left and right ventricular function (LVEF 53% and RVEF 52%), without segmental wall motion abnormalities. Of note, there was bilateral concentric hypertrophy and a circumferential subendocardial scar pattern consistent with cardiac amyloidosis (Figure 4).
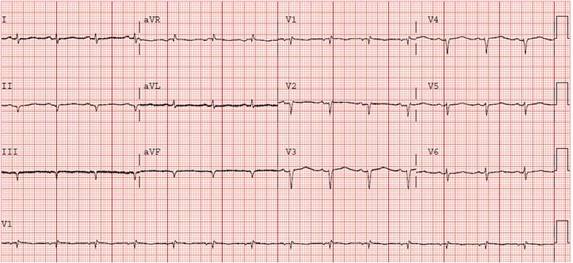
Figure 1. ECG showing low voltage throughout, pseudo-infarcts in the anterior and inferior leads, and poor R-wave progression.
Autopsy results revealed evidence of systemic amyloidosis, with vascular intraluminal deposition of amyloid involving the coronary arteries, liver, spleen, lung, thyroid, parathyroid, adrenal glands, testes, bone marrow, skeletal muscle, subcutaneous fat, and nerves. These deposits were homogenous eosinophilic material on H and E stains and Congo red stains, and demonstrated green birefringence under polarized light using a Thioflavin T stain (Figure 2). The cardiac deposits were further characterized as occurring in the interstitium between cardiac myocytes. Immunohistochemical stains were positive for IgG Kappa light chains and negative for IgG lambda light chains. The heart was grossly found to be hypertrophied, weighing 470g, with a right ventricle diameter 0.7cm, and a left ventricle diameter of 1.6cm. Thus, the patient's final diagnosis was amyloidosis secondary to monoclonal Kappa chain deposition by clonal B cell lymphoproliferative diseases (AL).
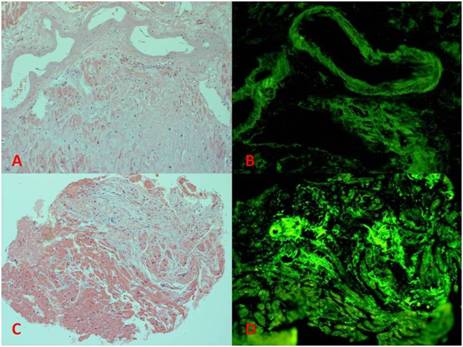
Figure 2. Showing H&E and Thioflavin T stains for cases 1 (images A and B) and 2 (images C and D). The H&E images highlight the eosinophilic cardiac myocytes with the amorphous amphophilic amyloid deposits in the perivascular and interstitial distributions. The Thioflavin T stains show the amyloid deposits in fluorescent green.
This is a 53 year old female without any known past medical history, who presented to our hospital with an 8 month history of progressive shortness of breath, paroxysmal nocturnal dyspnea, and lower extremity edema.
The patient exhibited atrial arrhythmias, specifically atrial fibrillation and atrial flutter, with rapid ventricular responses throughout her hospital course. She was treated with low dose beta-blockade with appropriate rate control. An echocardiogram was obtained which revealed normal left and right ventricular systolic function and cavity sizes, without associated wall motion abnormalities. There was severe left ventricular concentric hypertrophy with impaired left ventricular relaxation as evidenced by a reduced E/A ratio. There also was echocardiographic evidence of tamponade, which was demonstrated by an exaggerated mitral inflow variation (Figure 3).
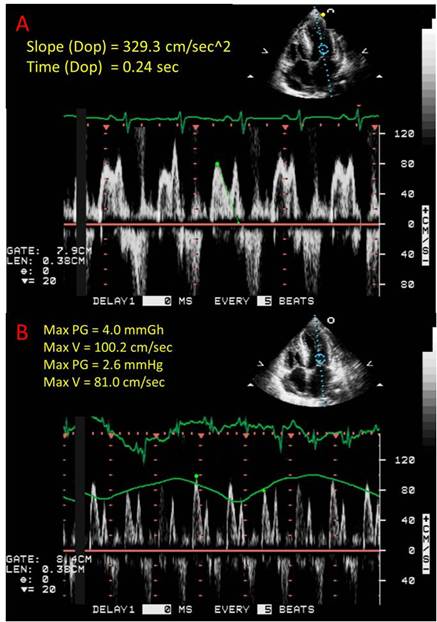
Figure 3. A) Echographicfindings suggestive of diastolic dysfunction as shown by an increased deceleration time (normal less than 220ms). B) Echographic evidence of tamponade characterized by excessive respiratory variations (normal <25%) in diastolic mitral valve inflow.
As the patient did not have a history of hypertension or aortic valve disease, concern over an infiltrative restrictive cardiomyopathy was evaluated. The patient had a normal serum and urine protein electrophoresis and normal serum light chains. She underwent an abdominal fat pad biopsy which did not stain with Congo red. Subsequent cardiac MRI with and without gadolinium was obtained, which was consistent with her previous echocardiograms (Figure 4). Additionally, MRI viability imaging was suggestive of an infiltrative process (not shown). The patient had an endomyocardial biopsy, which stained positively with Congo red, with green birefringence under polarized light (not shown) and positively for Thioflavin T (Figure 2). Immunohistochemistry staining was equivocal for transthyretin and negative for lambda and kappa light chains. Of note, the patient's cardiac biopsy was complicated by a supraventricular tachycardia, which was managed with transiently induced adenosine heart block, transvenous pacing, and intravenous metoprolol. She was subsequently monitored and discharged with appropriate beta blocker therapy.
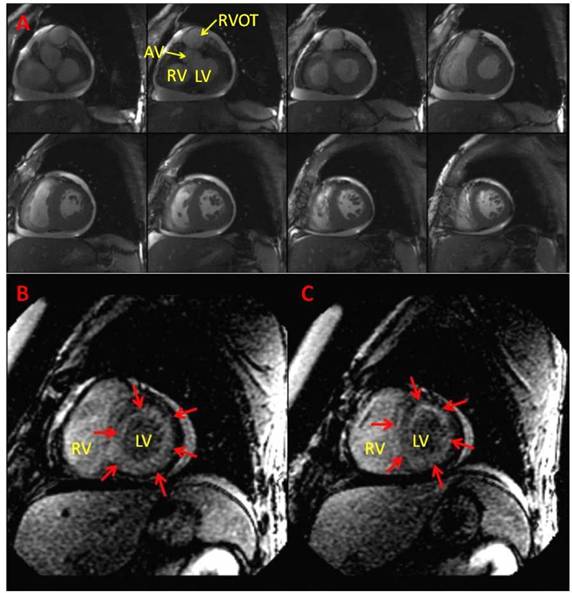
Figure 4. Cardiac Magnetic Resonance Imaging. (A) Steady-state free precession images of the basal and mid short axis views of LV at end diastole show biventricular concentric hypertrophy from case 2. (B, C) Delayed contrast-enhanced short-axis views of the LV demonstrating diffuse subendocardial enhancement (red arrows) from case 3. RV=Right Ventricle, LV=Left Ventricle, AV=Aortic valve, RVOT=Right ventricular outflow tract.
Given that the patient lacked a family history of cardiac diseases and did not have evidence of a chronic inflammatory state or plasma cell dyscrasia, the patient's cardiac amyloidosis was thought to be of the ATTRwt subtype. It is worthwhile to note that the patient's age is young for ATTRwt. A TTR gene sequencing test was not performed and that test could possibly have confirmed her as an ATTRm patient.
This is a 46 year old male with a family history of early cardiac death and known amyloidosis. He presented to our hospital with chest pain, worsening shortness of breath, swelling, and decreased exercise tolerance after a recent medication change; his furosemide was reduced from 80mg to 40mg orally twice daily. His diagnosis of amyloidosis was based on fat pad biopsy, which stained positive for transthyretin and kappa light chains. He went on to have an echocardiogram that showed a preserved left ventricular ejection fraction (60-65%), with concentric hypertrophy. This was followed by a nuclear medicine pyrophosphate study, showing diffuse pyrophosphate within the myocardium (Figure 5). Cardiac amyloidosis was suspected based on MRI, which showed a circumferential subendocardial (25-50%) perfusion defect, without preserved viability, consistent with cardiac amyloidosis (Figure 4).
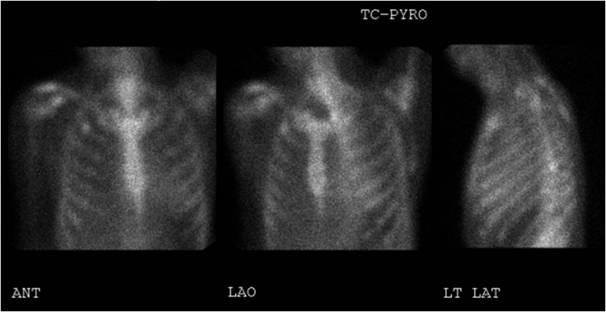
Figure 5. Nuclear medicine pyrophosphate study showing mildly diffuse pyrophosphate uptake within the myocardium which is less than adjacent skeletal uptake.
Cardiac amyloidosis had been previously confirmed when he underwent a cardiac catheterization five days prior to this presentation to obtain an endomyocardial tissue sample. The catheterization showed a thermodilution cardiac index of 2.2L/min/m2 (normal 2.6-4.2L/min/m2) and a pulmonary capillary wedge pressure of 24mmHg (normal 2-15mmHg). His biopsy resulted in positive Congo-red, Thioflavin T and amyloid P stains (Figure 6). Additionally, it had negative direct immunofluorescent stains for light chains and positive immunohistochemical stains for transthyretin. Given his family history of early cardiac death and his age, he was given a diagnosis of ATTRm.
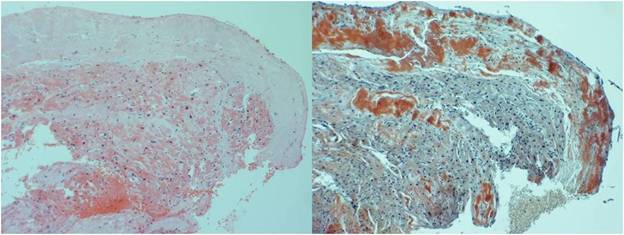
Figure 6. H & E (left) and Congo red stains (right) from case 3.
Unfortunately, while the patient was awaiting endomyocardial biopsy, he suffered a pulseless electrical activity arrest and died. The rhythm strip on telemetry prior to arrest showed a ventricular junctional rhythm.
During this hospital presentation, he was ruled out for acute coronary syndrome with serial electrocardiograms that were not suggestive of ischemia and negative troponins. Prior to discharge, he ambulated without symptoms and had negative orthostatic vital signs. He was subsequently discharged home on aspirin 81mg orally daily, carvedilol 25mg orally twice daily, furosemide 160mg orally daily, and simvastatin 20mg orally nightly.
Unfortunately, the patient returned to the emergency department the following day with weakness and dizziness. Upon presentation, his blood pressure was 65/41mmHg, with a heart rate of 75 beats per minute. His electrocardiogram showed a right bundle branch block and left anterior fascicular block. Shortly thereafter, the patient suffered a PEA arrest and despite prolonged standard advanced cardiovascular life support for 45 minutes, the patient died. Bedside ultrasound obtained during the resuscitation efforts did not show evidence of cardiac tamponade.
Clinical suspicion for cardiac amyloidosis should arise for any patient that presents with symptoms of restrictive cardiomyopathy. Unfortunately, these symptoms are not specific to cardiac amyloidosis. Symptoms include fatigue, exertional dyspnea, edema, coupled with appropriate physical exam and clinical data. Physical exam findings include macroglossia or indentations on the tongue (in AL), rales, pitting edema, elevated jugular venous pressure, and a third heart sound. Clinical data suggestive of cardiac amyloidosis include a low voltage ECG, or an ECG with atrial fibrillation or ventricular arrhythmias. Echocardiographic findings consistent with cardiac amyloidosis include concentric hypertrophy, a thickened interventricular septum, and diastolic dysfunction [1].
One recent cohort study by Murtagh et al. of cardiac amyloidosis patients showed that the most common ECG finding was low voltage on the limb leads (<5mm in height) and a pseudo-infarct pattern (pathologic Q waves in at least two consecutive leads) [8]. When both were present, there was a high specificity and positive predictive value for cardiac amyloidosis, of 98% and 96% respectively. Unfortunately, this combination also proved to be very insensitive, with a sensitivity of only 28% [5,6]. Not surprisingly, those diagnosed with cardiac amyloidosis via autopsy versus those diagnosed when alive, showed a lower percentage of low voltage ECGs (17.65% versus 66.67% respectively) [7]. The most common arrhythmia found was atrial fibrillation or atrial flutter, found in 12-20% of patients in two recent case series [5,8]. Nonspecific conduction delay was also a common finding in 16% of patients surveyed [8].
Classically amyloid deposition into the myocardium is described as a "sparkly" or granular pattern on echocardiography, which carries a greater than 80% sensitivity and specificity for cardiac amyloidosis [1,9]. Concentric ventricular thickening is often seen, with left ventricular thickening greater in ATTRwt [9]. Diastolic dysfunction is the earliest echocardiographic finding seen, as evidenced by high E wave and a low A wave indicating pseudonormal to severe diastolic dysfunction [2,7,9,10]. Newer echocardiographic techniques including tissue doppler imaging (TDI), TDI-derived strain, and 2D Strain or Speckle Tracking Imaging can show cardiac amyloidosis at a subclinical stage when other echocardiographic findings remain normal [9].
Nuclear medicine tracers using technetium pyrophosphate have been used to localize cardiac amyloidosis. This appears to occur with more sensitivity in transthyretina myloidosis subtypes (ATTRwt and ATTRm) versus the AL subtype [2].
Cardiovascular magnetic resonance (CMR) improves the diagnostic accuracy of cardiac amyloidosis, especially if it is enhanced with gadolinium. Late, global, subendocardial left ventricular enhancement with gadolinium is rarely seen in other cardiomyopathies and thus is very specific for cardiac amyloidosis [11].
The gold standard for the diagnosis of cardiac amyloidosis is an endomyocardial biopsy showing cardiac amyloid deposition. Endomyocardial biopsies are not required in patients with biopsy proven amyloidosis at other anatomical sites and have supportive echocardiographic or other noninvasive studies to suggest cardiac involvement. Biopsies are sometimes needed to differentiate underlying type of amyloidosis, which will in turn determine the appropriate treatment.
2021 Copyright OAT. All rights reserv
The mainstay of supportive treatment for cardiac amyloidosis revolves around salt restriction and diuretics. Indeed, many traditional heart failure treatments are relatively contraindicated. For instance, beta blockers are contraindicated for their negative inotropic properties. Both calcium channel blockers and digoxin can avidly bind to amyloid fibrils, leading to toxicity [12,13]. Since many patients have comorbid arrhythmias, amiodarone has emerged as an often used anti-arrhythmic. Unfortunately, there is not sufficient data to support the use of implantable defibrillators to prevent sudden cardiac death in cardiac amyloidosis patients [14].
Direct pharmacologic therapy for each type of amyloid is beyond the scope of this case series review. Briefly, we will address treatment directed towards AL, ATTRm, and ATTRwt amyloidosis as they commonly affect the heart.
In AL amyloidosis, treatments against plasma cells include combinations of chemotherapy consisting of dexamethasone, melphalan or other alkylators, lenalidomide, and/or bortezomib [15,16]. In patients with limited cardiac involvement, high dose chemotherapy prior to stem cell transplant is also an option in AL patients [15-17]. In select AL patients, i.e. those that can tolerate chemotherapy afterwards, cardiac transplantation can be beneficial [18].
In patients with ATTRm, the TTR protein is made in the liver and thus liver transplantation can be beneficial. Of note, certain TTR mutations will continue to show cardiac involvement despite liver transplantation and thus the Ala60 and non-Val30Met mutations tend to carry worse prognosis [19,20]. Clinical trials are underway to evaluate reducing TTR production by silencing RNA and antisense oligonucleotide therapies [21]. ATTRwt is treated symptomatically and patients with ATTRwt are often excluded from transplant given their advanced age. Theoretically, individuals with ATTRwt who are transplanted may continue to have misfolding of the wild-type transthyretin. However, anecdotally, younger ATTRwt patients have undergone cardiac transplantation successfully [1].
Prognosis varies based on the subtype of cardiac amyloidosis, with AL having a worse prognosis than TTR mutation subtypes (ATTRwt and ATTRm). Without treatment median survival for AL amyloidosis involving the heart is 6-12 months, although there is increasing evidence that with early appropriate treatment, survivability can extend past 5 years [22,23]. Cardiac AL patient’s prognosis is dependent on the severity and extension of organ involvement as well as response to therapy [24]. In a recent analysis of 102 biopsy proven ATTRwt cardiac amyloid patients and 36 AL cardiac amyloid patients, survival from diagnosis was significantly longer in the ATTRwt group (mean 2.71 years) versus the AL group (mean 0.87 years). Poorer survivability characteristics upon diagnosis of the ATTRwt group included a positive troponin, pacemaker placement, and higher NYHA class [25].
This case series highlights the three most common types of cardiac amyloidosis, namely AL, ATTRm, and ATTRwt. The common presenting symptoms where non-specific to cardiac amyloidosis but were more suggestive of heart failure in general. These included exertional chest pain, dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, and bilateral lower extremity swelling. Thus, a high level of clinical suspicion must be taken into account for any patients presenting with heart failure symptoms and other signs of systemic amyloidosis, i.e. renal dysfunction, abnormal protein/albumin ratios, or peripheral neuropathy. Although high clinical suspicion for cardiac involvement can be obtained through non-invasive techniques like echocardiography and cardiac magnetic resonance, tissue diagnosis via biopsy is still essential as the subtype of amyloidosis will dictate the disease management and prognosis.
The authors would like to thank Dr. Jina Chung for providing echocardiographic and cardiac MRI images. We would also like to thank our colleagues at Harbor-UCLA Medical Center for their support and guidance.
- Seldin DC, Berk JL, Sam F, Sanchorawala V (2011) Amyloidotic cardiomyopathy: Multidisciplinary approach to diagnosis and treatment. Heart Failure Clinics 7: 385-393. [Crossref]
- Esplin BL, Gertz MA (2013) Current trends in diagnosis and management of cardiac amyloidosis. Curr Probl Cardiol 38: 53-96. [Crossref]
- Sharma NH (2013) Current state of cardiac amyloidosis. Current Opinion in Cardiology 18: 157-162. [Crossref]
- Cornwell III GG, Murdoch WL, Kyle RA, Westermark P, Pitkänen P (1983) Frequency and distribution of senile cardiovascular amyloid: A clinicopathologic correlation. Am J Med 75: 618-623. [Crossref]
- Cheng Z, Zhu K, Tian Z, Zhao D, Cui Q, et al. (2013) The findings of electrocardiography in patients with cardiac amyloidosis. Annals of Noninvasive Electrocardiology 18: 157-162. [Crossref]
- Kyle R, Linos A, Beard C, Linke R, Gertz M, et al. (1992) Incidence and natural history of primary systemic amyloidosis in olmsted county, minnesota, 1950 through 1989 [see comments]. Blood 79: 1817-1822. [Crossref]
- Gutierrez PS, Fernandes F, Mady C, Higuchi ML (2008) Clinical, electrocardiographic and echocardiographic findings in significant cardiac amyloidosis detected only at necropsy: Comparison with cases diagnosed in life. Arq Bras Cardiol 90: 191-196. [Crossref]
- Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, et al. (2005) Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 95: 535-537. [Crossref]
- Tsang W, Lang R (2010) Echocardiographic evaluation of cardiac amyloid. Curr Cardiol Rep 12: 272-276. [Crossref]
- Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD (2012) Updates in cardiac amyloidosis: A review. J Am Heart Assoc 1: e000364. [Crossref]
- Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, et al. (2008) Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: Noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol 51: 1022-1030. [Crossref]
- Rubinow A, Skinner M, Cohen AS (1981) Digoxin sensitivity in amyloid cardiomyopathy. Circulation 63: 1285-1288. [Crossref]
- Yamaguchi T (2008) Syncope and sinus bradycardia from combined use of thalidomide and B-blocker. Pharmacoepidemiol Drug Saf 17: 1033-1035. [Crossref]
- Kristen AV, Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, et al. (2008) Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm 5: 235-240. [Crossref]
- Falk RH (2011) Cardiac amyloidosis: A treatable disease, often overlooked. Circulation 124: 1079-1085. [Crossref]
- Meier-Ewert H, Sanchorawala V, Berk J, Ruberg F (2011) Cardiac amyloidosis: Evolving approach to diagnosis and management. Curr Treat Options Cardiovasc Med 13: 528-542. [Crossref]
- Gertz MA, Lacy MQ, Dispenzieri A, Gastineau DA, Chen MG, et al. (2002) Stem cell transplantation for the management of primary systemic amyloidosis. Am J Med 113: 549-555. [Crossref]
- Sattianayagam PT, Gibbs SDJ, Pinney JH, Wechalekar AD, Lachmann HJ, et al. (2010) Solid organ transplantation in AL amyloidosis. Am J Transplant 10: 2124-2131. [Crossref]
- Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SDJ, Pinney JH, et al. (2012) Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J 33: 1120-1127. [Crossref]
- Liepnieks JJ, Benson MD (2007) Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid 14: 277-282. [Crossref]
- Benson MD, Kluve-Beckerman B, Zeldenrust SR, Siesky AM, Bodenmiller DM, et al. (2006) Targeted suppression of an amyloidogenictransthyretin with antisense oligonucleotides. Muscle Nerve 33: 609-618. [Crossref]
- Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, et al. (2009) Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 120: 1203-1212. [Crossref]
- Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN (2012) Cardiac transthyretin amyloidosis. Heart 98: 1546-1554. [Crossref]
- Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, et al. (2013) Cardiac amyloidosis: Updates in diagnosis and management. Arch Cardiovasc Dis 106: 528-540. [Crossref]
- Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, et al. (2013) Senile systemic amyloidosis: Clinical features at presentation and outcome. J Am Heart Assoc 2: e000098. [Crossref]