Abstract
Cancer is the second most common cause of death in the world, and primary prevention remains the best approach to reducing overall morbidity and mortality. There is a molecular link between cyclooxygenase-2 (COX-2)-derived prostaglandin E2 (PGE2) production and colonic tumorigenesis. Selective COX-2 inhibitors as well as non-steroidal anti-inflammatory drugs (NSAIDs) reduce the number and sizes of colonic adenomas; however, increased cardiovascular risks of selective COX-2 inhibitors and increased gastrointestinal side-effects of NSAIDs limit their use. Glucocorticoids induce cancer cell apoptosis and are endogenous, potent COX-2 inhibitors. In tissues, glucocorticoid actions are down-regulated by type 2 11ß-hydroxysteroid dehydrogenase (11ßHSD2), and inhibition of 11ßHSD2 activity will elevate intracellular active glucocorticoids to levels that effectively suppress COX-2 expression. Both COX-2 and 11ßHSD2 increase in Apc+/min mouse intestinal adenomas and human colonic adenomas, and either pharmacologic or genetic 11βHSD2 inhibition leads to decreases in COX-2–mediated PGE2 production in tumors and prevents adenoma formation, tumor growth, and metastasis. 11ßHSD2 inhibition may represent a novel approach for CRC chemoprevention by increasing tumor cell intracellular glucocorticoid activity, which in turn inhibits tumor growth by suppressing the COX-2-derived PGE2 pathway, as well as other pathways, without potential side-effects relating to chronic application of COX-2 inhibitors, NSAIDs and glucocorticoids.
Key words
11ßHSD2, cyclooxygenase-2, glucocorticoid, cancer
Introduction
Cancers are leading causes of morbidity and mortality worldwide, with approximately 14 million new cases and 8.2 million cancer related deaths per year after 2012. The number of new cases is expected to rise by about 70% over the next 2 decades [1]. Primary prevention remains the best approach to reducing overall morbidity and mortality.
The etiology of cancer is undoubtedly multifactorial, but there is experimental and clinical evidence linking abnormalities in the cyclooxygenase/prostaglandin system to its pathogenesis. Cyclooxygenase (prostaglandin synthase G2/H2, COX) is the rate-limiting enzyme in the metabolism of arachidonic acid to prostaglandin G2 and subsequently to prostaglandin H2 (PGH2), which serves as the precursor for prostaglandin E synthetase to produce prostaglandins [2]. Two isoforms of cyclooxygenase exist in mammals, COX-1 and COX-2. COX-2 derived PGE2 has been reported to promote tumor growth through stimulation of cell proliferation, cell migration, cell invasion, angiogenesis and immunosuppression [3].
Cyclooxygenase-2 and glucocorticoids in tumorigenesis
COX-2 was initially described as an inflammatory-mediated cyclooxygenase. COX-2 plays an important role in tumor cell growth and in cancer metastasis [4-7]. Hepatic metastases in colorectal cancer (CRC) develop more frequently when tumors express high COX-2 levels. COX-2 expression increases not only in the CRC and lung cancer, but to even higher levels in hepatic metastases [4].
COX-2 expression is sensitive to glucocorticoid inhibition. Glucocorticoids (GCs) are the most potent, endogenous, specific COX-2 inhibitors, acting to suppress COX-2 expression through stimulating glucocorticoid receptors [8-10]. Glucocorticoids are produced in the zona fasciculata (middle cortical layer) of the adrenal gland. Activation of glucocorticoid receptors inhibits COX-2 expression primarily through three mechanisms: 1. Transcriptional inhibition. Activation of either activator protein-1 (AP-1) or nuclear factor- κB (NF-κB) leads to increased COX-2 transcription. GCs inhibit COX-2 expression by direct interaction with AP-1 and NF-κB. 2. Post-transcriptional inhibition. Sustained p38 activity is required for the stabilization of COX-2 mRNA. MAPK phosphatase-1 (MKP-1), the founding member of MAPKs family, which plays an important role in cell proliferation, apoptosis and immune defense, is induced by GCs and inactivates pp-38, leading to destabilization of COX-2 mRNA. 3. Indirect transcriptional inhibition. In addition to the transcriptional inhibition, GCs also induce the expression of several proteins that suppress COX-2 expression such as IκBα, which directly inhibits NF-κB activity [11]. GCs can also inhibit COX-2 expression through induction of lipocortin-1, which suppresses phospholipase A2 and inhibits leukocyte inflammatory events (epithelial adhesion, emigration, chemotaxis, phagocytosis, etc). In addition to inhibiting COX-2 expression, GCs reduce PG production through inhibition of cytosolic PLA 2 (cPLA2) activity, which prevents the release of arachidonic acid from membrane phospholipids, and through inhibition of microsomal prostaglandin E synthetase (mPGES-1) expression, a major terminal synthetase in PGE2 biosynthesis [12]. mPGES-1 and COX-2 expression are significantly increased in human colorectal tumors [12,13]. Local tissue levels of PGE 2 are determined by PGE2 biosynthesis and degradation. The NAD+-dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH) catalyzes the rate-limiting step of PG catabolism, and 15-PGDH expression decreases in CRC [14]. GCs induce 15-PGDH expression in A549 human lung adenocarcinoma cells [15]. Therefore, GCs may reduce tissue levels of PGs through coordinated inhibition of cPLA2 activity, COX-2 and mPGES-1 expression, and induction of 15-PGDH.
GCs have been used to treat diseases caused by an overactive immune system, such as allergies, asthma, and autoimmune diseases as well as a broad spectrum of hematologic malignancies, including leukemia, lymphoma, and myeloma due to their ability to induce apoptosis [16-18]. Recent studies found the inhibitory effect of GCs to COX-2 could suppress solid tumor growth, regress tumor mass, and prevent metastasis by blocking angiogenesis [19,20]. However, the undesirable side effects of immunosuppression limit their application in cancer chemoprevention and chemotherapy.
Circulating glucocorticoid levels are regulated by the hypothalamo-pituitary-adrenal axis [21]. Peptide corticotropin-releasing hormone (CRH) released from the hypothalamus following stressors stimulates the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, which in turn stimulates the adrenal gland to secrete glucocorticoids. When blood concentrations of glucocorticoids rise above a certain threshold, CRH secretion is inhibited from the hypothalamus, leading to turning off of ACTH secretion and subsequent turning off of glucocorticoid secretion from the adrenal gland. In addition to systemic regulation, glucocorticoid effects are also regulated locally in tissues by a “pre-receptor” regulatory mechanism involving 11ß-hydroxysteroid dehydrogenase type I (11ßHSD1) and 11ßHSD2 [22]. 11ßHSD1 produces active glucocorticoids from inactive metabolites, while 11ßHSD2 converts glucocorticoids to their inactive ketoforms. 11ßHSD1 is expressed in key glucocorticoid target tissues such as liver and adipose tissue, while 11ßHSD2 is expressed predominantly in mineralocorticoid responsive tissues such as the kidney, salivary gland and colon [23-26].
11ßHSD2 acts as a procarcinogenic agent through attenuation of glucocorticoid actions and suppresses tumorigenesis through enhancing glucocorticoid-mediated inhibition of COX-2 pathway
11ßHSD2 has been proposed to be a pro-proliferative agent due to its ability to attenuate the anti-proliferative effects of glucocorticoids. The most conclusive evidence for the role of 11ßHSD2 in tumorigenesis comes from the analysis of 11ßHSD isoenzyme expression in a large cohort of human pituitary tumors [27-29]. 11ßHSD1 expression decreases, while 11ßHSD2 expression dramatically increases in pituitary tumors compared with normal tissues. Inhibition of 11ßHSD2 activity suppresses pituitary cell proliferation and augments glucocorticoid anti-proliferative effects. 11ßHSD2 expression increases in human adrenal cortical carcinoma/adenoma, breast cancers, and neoplastic cell lines [30-35]. In cultured cell lines, overexpression of 11ßHSD2 stimulates cell proliferation, while inhibition of 11ßHSD2 activity suppresses cell proliferation [31-34].
11ßHSD2 activity is higher in the colon than in the small intestine in the rat and it is primarily localized to intestinal epithelia [36]. In intestinal tumors, 11ßHSD2 is expressed in epithelial and stromal cells [37-39]. Interestingly, the incidence of intestinal tumors is higher in the colon than in the small intestine. The mechanism underlying increases in COX-2 expression in CRC is not completely understood. Recent studies have found that 11ßHSD2 mRNA levels are significantly higher in both human colonic small and large adenomas compared to adjacent normal colon tissues [40]. Immunohistochemical staining showed that increased 11ßHSD2 expression is primarily localized to adenoma epithelia and co-localized with increased COX-2 expression in human colonic adenomas. Both 11ßHSD2 and COX-2 expression also increase in adenomas of Apc+/min mice. Mouse adenocarcinoma CT26 cells demonstrate significant tumorigenic activity and express high levels of COX-2, and both their proliferation in vitro and the size and number of CT26 tumors in vivo are sensitive to COX-2 inhibition [40]. CT26 cells also express 11ßHSD2. Corticosterone treatment (the active glucocorticoid in rodents) leads to inhibition of CT26 cell COX-2 expression, which is enhanced by pharmacologic or genetic inhibition of 11ßHSD2 activity but is attenuated by overexpression of 11ßHSD2. Pharmacologic inhibition of 11ßHSD2 activity with glycyrrhizic acid (GA) suppresses adenoma development and growth, associated with inhibition of adenoma COX-2 and mPGES-1 expression in Apc+/min mice. Knockdown of 11ßHSD2 in CT26 cells leads to inhibition of CT26 tumor growth associated with inhibition of CT26 tumor COX-2 and mPGES-1 expression as well as PGE2 production. Pharmacologic inhibition of 11ßHSD2 activity with GA increases active glucocorticoid levels but decreases inactive 11-keto-corticosterone levels in CT26 tumors as well as in tissues with high levels of 11ßHSD2 activity such as kidney and colon. GA treatment leads to decreases in CT26 tumor incidence, inhibition of CT26 tumor growth, decreases in tumor phosphorylated cPLA2 (active form), COX-2 and mPGES-1 expression as well as tumor PGE2 levels; all of these parameter are reversed by treatment with RU486, an inhibitor of glucocorticoid receptors. Human colon carcinoma HCA-7 cells constitutively express both 11ßHSD2 and COX-2, and COX-2 activity is essential for the proliferation of HCA-7 cells [41]. On the other hand, human colon carcinoma HT-29 cells constitutively express similar levels of 11ßHSD2 with minimal COX-2 expression, and COX-2 activity is not essential for HT-29 cell proliferation [42]. Inhibition of 11ßHSD2 activity with glycyrrhizic acid has no effect on HT-29 tumor growth, but significantly inhibits HCA-7 tumor growth, which is associated with decreases in tumor COX-2 and mPGES-1 expression. Therefore, 11ßHSD2 inhibition represents a novel approach for colorectal cancer chemoprevention and therapy by increasing tumor glucocorticoid activity, which in turn selectively blocks local COX-2 activity.
In addition to inhibition of the COX-2 pathway, increased intracellular glucocorticoids due to 11ßHSD2 inhibition may suppress proliferation and induce apoptosis and differentiation through other mechanisms. The retinoblastoma protein (Rb), a tumor suppressor, inhibits cell proliferation through its interaction with the E2F family of transcription factors and the resultant regression of genes that are essential for DNA synthesis [43]. The inactivation of retinoblastoma protein is a prerequisite for cell proliferation. Inactivation of Rb is achieved through cyclin-dependent protein kinase-mediated phosphorylation during cell cycle progression. Glucocorticoids inactivate Rb through p53-mediated induction of p21waf1/cip1, an inhibitor of cyclin-dependent kinase [44-48]. Glucocorticoids may also inhibit tumor growth through suppression of the mTOR signal pathway [49,50]. REDD1 (RTP801, an mTOR complex 1, mTORC1, repressor) is a novel stress-induced gene linked to regression of mTOR signaling and is induced by glucocorticoids [51-53]. Recently, we generated Apc+/min mice with selective deletion of 11ßHSD2 in intestinal epithelial cells, and intestinal tumorigenesis is inhibited in these mice. In adenomas from these mice, p53 and p21 levels increased, with concomitant decrease in phosphorylation of retinoblastoma protein. In addition, adenoma REDD1 increased, while the activity of mTOR signaling pathway was inhibited.
The lipoxygenase (LOX) pathway is another important pathway for metabolism of arachidonic acid. 5-LOX and its metabolite, leukotriene B4, have been reported to stimulate the proliferation of several human colon carcinoma cell lines [54,55]. 5-LOX increases in human colorectal tumors, and inhibition of the 5-LOX pathway suppresses the proliferation of human colon carcinoma cell lines and inhibits CRC development [56-60]. Dexamethasone has been reported to inhibit 5-LOX expression and its metabolite production [61,62].
Conclusion and future perspective
The inhibition of 11βHSD2 activity may provide a new target for chemoprevention and/or adjunctive therapy for cancers (CRC, lung cancer, etc), particularly for patients with increased risk, such as familial adenomatous polyposis (FAP) patients, as a result of increased tumor intracellular active glucocorticoids, because of the following advantages (Figure 1): 1. Glucocorticoids selectively inhibit COX-2, but not COX-1, mediated prostaglandin production. Therefore, inhibition of 11βHSD2 activity has the beneficial effects of traditional NSAIDs to prevent and regress colorectal cancers without the gastrointestinal side effects associated with COX-1 inhibition; 2. Physiologic 11βHSD2 is most highly expressed in colon and kidney. Therefore, inhibition of 11βHSD2 activity is not expected to incur the cardiovascular risk posed by COX-2 inhibitors that suppress COX-2–derived PGI2 production in vascular endothelial cells; 3. Increased levels of intracellular active glucocorticoids are observed only in tissues with elevated 11βHSD2 expression. Inhibition of 11βHSD2 will not produce immunosuppression or other systemic side effects of conventional glucocorticoid therapy; 4. Increased tumor active glucocorticoids also inhibit tumor development and growth through induction of G1 cell cycle arrest and inhibition of the mTOR pathway; 5. Tissue prostaglandin levels are determined by both biosynthesis and catabolism. A key enzyme in prostaglandin catabolism, 15-hydroxyprostaglandin has been reported to be decreased in Apc+/min mouse intestinal adenomas and in human colorectal cancer [63], and glucocorticoids have been shown to induce 15-hydroxyprostaglandin in A549 human lung adenocarcinoma cells [64]. Therefore, it is possible that increased intracellular active endogenous glucocorticoids may inhibit tumorigenesis not only by inhibiting prostaglandin synthesis, but also by enhancing prostaglandin degradation; and finally 6. Glucocorticoid also inhibit the activity of cPLA2, preventing potential shunting of arachidonic acid metabolism to other procarcinogenic metabolizing pathway such as the 5-lipoxygenase lipoxygenase pathway [60-62,65]. In addition, glucocorticoids inhibit both COX-2 and 5-LOX.
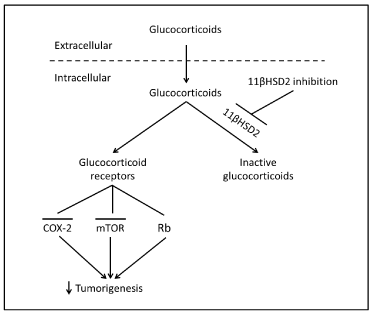
Figure 1. Proposed mechanism underlying epithelial cell 11ßHSD2 activity and tumorigenesis. In tumor epithelial cells, glucocorticoids are converted to inactive 11-keto-forms, reducing glucocorticoid receptor activity, while 11ßHSD2 inhibition leads to increased levels of epithelial intracellular active glucocorticoid. The subsequent inhibition of the COX-2 pathway and induction of G1 cell cycle arrest through activation of retinoblastoma protein (Rb, a tumor suppressor) and inhibition of the mTOR pathway lead to inhibition of colorectal tumorigenesis.
Glycyrrhizic acid and its analogs are excellent prototypes for 11βHSD2 inhibitors. It is a natural compound contained in licorice, a natural botanical antiinflammatory agent and a powerful 11βHSD2 inhibitor. Long-term excessive ingestion of licorice has been reported to induce hypokalemia and elevation of blood pressure in a subset of people [66]. Although these side effects were not seen in animal experiments, studies did observe that the concentrations of glycyrrhizic acid increased levels of active glucocorticoid levels in the kidney [40]. Although 11βHSD2 inhibition may result in salt-dependent hypertension due to activation of mineralocorticoid receptor by glucocorticoids in kidney, development of locally acting enteric 11ßHSD2 inhibitors that are not systemically absorbed would be a potential therapeutic means to prevent tumorigenesis [67].
Acknowledgments
This work was supported by CA-122620, DK-95785, DK-51265, DK-62794, and by funds from the Department of Veterans Affairs (RCH).
References
- World Cancer Report 2014.
- Frankfurt O, Rosen ST (2004) Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol 16: 553-563. [Crossref]
- Almawi WY, Abou Jaoude MM, Li XC (2002) Transcriptional and post-transcriptional mechanisms of glucocorticoid antiproliferative effects. Hematol Oncol 20: 17-32. [Crossref]
- Chen WS, Wei SJ, Liu JM, Hsiao M, Kou-Lin J, et al. (2001) Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer 91: 894-899. [Crossref]
- Kakiuchi Y, Tsuji S, Tsujii M, Murata H, Kawai N, et al. (2002) Cyclooxygenase-2 activity altered the cell-surface carbohydrate antigens on colon cancer cells and enhanced liver metastasis. Cancer Res 62: 1567-1572. [Crossref]
- Soumaoro LT, Uetake H, Higuchi T, Takagi Y, Enomoto M, et al. (2004) Cyclooxygenase-2 expression: a significant prognostic indicator for patients with colorectal cancer. Clin Cancer Res 10: 8465-8471. [Crossref]
- Tsujii M, Kawano S, DuBois RN (1997) Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A 94: 3336-3340. [Crossref]
- Clark AR, Lasa M (2003) Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr Opin Pharmacol 3: 404-411. [Crossref]
- Newton R (2000) Molecular mechanisms of glucocorticoid action: what is important? Thorax 55: 603-613. [Crossref]
- Zhang MZ, Harris RC, McKanna JA (1999) Regulation of cyclooxygenase-2 (COX-2) in rat renal cortex by adrenal glucocorticoids and mineralocorticoids. Proc Natl Acad Sci U S A 96: 15280-15285. [Crossref]
- Newton R (2000) Molecular mechanisms of glucocorticoid action: what is important? Thorax 55: 603-613. [Crossref]
- Stichtenoth DO, Thorén S, Bian H, Peters-Golden M, Jakobsson PJ, et al. (2001) Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J Immunol 167: 469-474. [Crossref]
- Zhang MZ, Xu J, Yao B, Yin H, Cai Q, et al. (2009) Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest 119: 876-885. [Crossref]
- Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, et al. (2005) 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem 280: 3217-3223. [Crossref]
- Tong M, Tai HH (2005) 15-Hydroxyprostaglandin dehydrogenase can be induced by dexamethasone and other glucocorticoids at the therapeutic level in A549 human lung adenocarcinoma cells. Arch Biochem Biophys 435: 50-55. [Crossref]
- Frankfurt O, Rosen ST (2004) Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol 16: 553-563. [Crossref]
- Almawi WY, Abou Jaoude MM, Li XC (2002) Transcriptional and post-transcriptional mechanisms of glucocorticoid antiproliferative effects. Hematol Oncol 20: 17-32. [Crossref]
- Sionov RV, Spokoini R, Kfir-Erenfeld S, Cohen O, Yefenof E (2008) Mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoid-induced apoptosis. Adv Cancer Res 101: 127-248. [Crossref]
- Keith BD (2008) Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 8: 84. [Crossref]
- Yemelyanov A, Czwornog J, Chebotaev D, Karseladze A, Kulevitch E, et al. (2007) Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene 26: 1885-1896. [Crossref]
- Stewart PM, Prescott SM (2009) Can licorice lick colon cancer? J Clin Invest 119: 760-763. [Crossref]
- Funder JW, Pearce PT, Smith R, Smith AI (1988) Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242: 583-585. [Crossref]
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, et al. (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294: 2166-2170. [Crossref]
- Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, et al. (1999) Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest 103: 683-689. [Crossref]
- Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, et al. (1997) 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A 94: 14924-14929. [Crossref]
- Moore XL, Hoong I, Cole TJ (2000) Expression of the 11beta-hydroxysteroid dehydrogenase 2 gene in the mouse. Kidney Int 57: 1307-1312. [Crossref]
- Rabbitt EH, Ayuk J, Boelaert K, Sheppard MC, Hewison M, et al. (2003) Abnormal expression of 11 beta-hydroxysteroid dehydrogenase type 2 in human pituitary adenomas: a prereceptor determinant of pituitary cell proliferation. Oncogene 22: 1663-1667.
- Rabbitt EH, Gittoes NJ, Stewart PM, Hewison M (2003) 11beta-hydroxysteroid dehydrogenases, cell proliferation and malignancy. J Steroid Biochem Mol Biol 85: 415-421. [Crossref]
2021 Copyright OAT. All rights reserv
- Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Stewart PM, et al. (2002) Prereceptor regulation of glucocorticoid action by 11beta-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J 16: 36-44. [Crossref]
- Coulter CL, Smith RE, Stowasser M, Sasano H, Krozowski ZS, et al. (1999) Expression of 11beta-hydroxysteroid dehydrogenase type 2 (11betaHSD-2) in the developing human adrenal gland and human adrenal cortical carcinoma and adenoma. Mol Cell Endocrinol 154: 71-77. [Crossref]
- Hundertmark S, Buhler H, Rudolf M, Weitzel HK, Ragosch V (1997) Inhibition of 11 beta-hydroxysteroid dehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF-7 and ZR-75-1 breast cancer cells. J Endocrinol 155: 171-180. [Crossref]
- Lipka C, Mankertz J, Fromm M, Lübbert H, Bühler H, et al. (2004) Impairment of the antiproliferative effect of glucocorticosteroids by 11beta-hydroxysteroid dehydrogenase type 2 overexpression in MCF-7 breast-cancer cells. Horm Metab Res 36: 437-444. [Crossref]
- Koyama K, Krozowski Z (2001) Modulation of 11 beta-hydroxysteroid dehydrogenase type 2 activity in Ishikawa cells is associated with changes in cellular proliferation. Mol Cell Endocrinol 183: 165-170. [Crossref]
- Koyama K, Myles K, Smith R, Krozowski Z (2001) Expression of the 11beta-hydroxysteroid dehydrogenase type II enzyme in breast tumors and modulation of activity and cell growth in PMC42 cells. J Steroid Biochem Mol Biol 76: 153-159. [Crossref]
- Takahashi K, Sasano H, Fukushima K, Hirasawa G, Miura H, et al. (1998) 11 beta-hydroxysteroid dehydrogenase type II in human colon: a new marker of fetal development and differentiation in neoplasms. Anticancer Res 18: 3381-3388. [Crossref]
- Smith RE, Maguire JA, Stein-Oakley AN, Sasano H, Takahashi K, et al. (1996) Localization of 11 beta-hydroxysteroid dehydrogenase type II in human epithelial tissues. J Clin Endocrinol Metab 81: 3244-3248. [Crossref]
- Takahashi K, Sasano H, Fukushima K, Hirasawa G, Miura H, et al. (1998) 11 beta-hydroxysteroid dehydrogenase type II in human colon: a new marker of fetal development and differentiation in neoplasms. Anticancer Res 18: 3381-3388. [Crossref]
- Whorwood CB, Ricketts ML, Stewart PM (1994) Epithelial cell localization of type 2 11 beta-hydroxysteroid dehydrogenase in rat and human colon. Endocrinology 135: 2533-2541. [Crossref]
- Zbánková S, Bryndová J, Kment M, Pácha J (2004) Expression of 11beta-hydroxysteroid dehydrogenase types 1 and 2 in colorectal cancer. Cancer Lett 210: 95-100. [Crossref]
- Zhang MZ, Xu J, Yao B, Yin H, Cai Q, et al. (2009) Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest 119: 876-885. [Crossref]
- Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, et al. (1997) Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest 99: 2254-2259. [Crossref]
- Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S (2002) Selective cyclooxygenase-2 inhibitors show a differential ability to inhibit proliferation and induce apoptosis of colon adenocarcinoma cells. FEBS Lett 531: 278-284. [Crossref]
- Chau BN, Wang JY (2003) Coordinated regulation of life and death by RB. Nat Rev Cancer 3: 130-138. [Crossref]
- Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, et al. (1994) Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest 94: 2504-2510. [Crossref]
- Cram EJ, Ramos RA, Wang EC, Cha HH, Nishio Y, et al. (1998) Role of the CCAAT/enhancer binding protein-alpha transcription factor in the glucocorticoid stimulation of p21waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J Biol Chem 273: 2008-2014. [Crossref]
- Cha HH, Cram EJ, Wang EC, Huang AJ, Kasler HG, et al. (1998) Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J Biol Chem 273: 1998-2007. [Crossref]
- Reil TD, Kashyap VS, Sarkar R, Freishlag J, Gelabert HA (2000) Dexamethasone inhibits the phosphorylation of retinoblastoma protein in the suppression of human vascular smooth muscle cell proliferation. J Surg Res 92: 108-113. [Crossref]
- Addeo R, Casale F, Caraglia M, D'Angelo V, Crisci S, et al. (2004) Glucocorticoids induce G1 arrest of lymphoblastic cells through retinoblastoma protein Rb1 dephosphorylation in childhood acute lymphoblastic leukemia in vivo. Cancer Biol Ther 3: 470-476. [Crossref]
- Polman JA, Hunter RG, Speksnijder N, van den Oever JM, Korobko OB, et al. (2012) Glucocorticoids Modulate the mTOR Pathway in the Hippocampus: Differential Effects Depending on Stress History. Endocrinology 153: 4317-4327. [Crossref]
- Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, et al. (2011) Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 13: 170-182. [Crossref]
- Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR (2006) Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem 281: 39128-39134. [Crossref]
- Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, et al. (2011) Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem 286: 30181-30189. [Crossref]
- Kumari R, Willing LB, Jefferson LS, Simpson IA, Kimball SR (2011) REDD1 (regulated in development and DNA damage response 1) expression in skeletal muscle as a surrogate biomarker of the efficiency of glucocorticoid receptor blockade. Biochem Biophys Res Commun 412: 644-647. [Crossref]
- Qiao L, Kozoni V, Tsioulias GJ, Koutsos MI, Hanif R, et al. (1995) Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochim Biophys Acta 1258: 215-223. [Crossref]
- Bortuzzo C, Hanif R, Kashfi K, Staiano-Coico L, Shiff SJ, et al. (1996) The effect of leukotrienes B and selected HETEs on the proliferation of colon cancer cells. Biochim Biophys Acta 1300: 240-246. [Crossref]
- Ohd JF, Nielsen CK, Campbell J, Landberg G, Löfberg H, et al. (2003) Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology 124: 57-70. [Crossref]
- Barresi V, Grosso M, Vitarelli E, Tuccari G, Barresi G (2007) 5-Lipoxygenase is coexpressed with Cox-2 in sporadic colorectal cancer: a correlation with advanced stage. Dis Colon Rectum 50: 1576-1584. [Crossref]
- Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, et al. (2006) Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther 5: 2716-2726. [Crossref]
- Ye YN, Wu WK, Shin VY, Bruce IC, Wong BC, et al. (2005) Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 26: 827-834. [Crossref]
- Jame AJ, Lackie PM, Cazaly AM, Sayers I, Penrose JF, et al. (2007) Human bronchial epithelial cells express an active and inducible biosynthetic pathway for leukotrienes B4 and C4. Clin Exp Allergy 37: 880-892. [Crossref]
- Peters-Golden M, Sampson AP (2003) Cysteinyl leukotriene interactions with other mediators and with glucocorticosteroids during airway inflammation. J Allergy Clin Immunol 111: S37-S48. [Crossref]
- Torosyan Y, Dobi A, Naga S, Mezhevaya K, Glasman M, et al. (2006) Distinct effects of annexin A7 and p53 on arachidonate lipoxygenation in prostate cancer cells involve 5-lipoxygenase transcription. Cancer Res 66: 9609-9616. [Crossref]
- Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, et al. (2005) 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem 280: 3217-3223. [Crossref]
- Tong M, Tai HH (2005) 15-Hydroxyprostaglandin dehydrogenase can be induced by dexamethasone and other glucocorticoids at the therapeutic level in A549 human lung adenocarcinoma cells. Arch Biochem Biophys 435: 50-55. [Crossref]
- Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, et al. (2006) Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther 5: 2716-2726. [Crossref]
- Størmer FC, Reistad R, Alexander J (1993) Glycyrrhizic acid in liquorice--evaluation of health hazard. Food Chem Toxicol 31: 303-312. [Crossref]
- Stewart PM, Prescott SM (2009) Can licorice lick colon cancer? J Clin Invest 119: 760-763. [Crossref]